


【超级大单】“面板双雄”遭抛弃!大单频出手一类股(附赠一图看懂碳中和机会)
【主力资金】炸裂8连板!连续两日尾盘出现神奇走势(附赠数字人民币受益股)
来源:崇利论市
粤开证券研究院首席策略分析师 陈梦洁
执业编号:S0300520100001
摘要
优选靶点,避免扎堆。随着DS-8201革命性的成功,自2020年起,ADC药物呈现出资本竞逐的局面。截至2021年3月,全球已有十款ADC药物获批上市,其中Kadcyla和Adcetris均于2020年在我国获批上市。从研发的角度,HER2 ADC的研发管线已日渐拥挤,而T-DM1的先发优势和DS-8201的优异数据则进一步加剧了药物上市后的风险。从研发的角度,扎堆适应症“跟跑”的价值十分有限,走差异化的创新之路是企业弯道超车的捷径。布局ADC药物的企业应另辟蹊径,遴选“人迹罕至”的靶点和适应症加速创新,抢滩登陆,发挥先发优势。
ADC产品涉及的技术呈现出高度的多样性和复杂性,差异化的设计会带来全新的产品。通过梳理已上市ADC产品的特点,我们抽丝剥茧,总结成功ADC产品的共性与发展趋势:
抗体:抗体经历了从鼠源抗体、人鼠嵌合单抗到人源化抗体的转变,逐步克服了人抗鼠抗体反应;IgG1抗体的广泛使用,增强了ADCC和CDC作用;抗体亲和力不断提升,不良反应发生有望下降。
Linker:从不可剪切Linker到可剪切Linker,再到DolaLock Payload,旁观者效应(Bystander effect)从无到有,从有到优,更好地发挥旁杀伤作用;从随机偶联到定点偶联,ADC药物的均一性逐步提升,但考虑到至今尚无定点偶联ADC药物获批上市,后续仍需持续关注;使用聚乙二醇(PEG)连接物调整水油分布系数,改善ADC药物亲水性,可能有助于对抗ADC耐药。
Payload:尽管微管蛋白抑制剂使用较广,但与DNA抑制剂相比,存在半衰期较长、抗癌谱较窄,靶点数量较多等弊端,正逐步被DNA抑制剂取代。新一代ADC药物凭借中等毒性的Payload搭配高DAR的设计,不仅可以更好地发挥抗肿瘤作用,而且脱靶毒性有望降低。Mersana公司的Fleximer平台目前已经实现DAR高达10-12的ADC药物,可进一步改善效应分子的运输效率。
受全球创新浪潮的冲击和ADC药物的更新迭代,国内ADC药物的研发热情高涨,一批生物医药企业相继奔赴ADC药物创新的战场。除了荣昌生物、浙江医药等重点布局HER2 ADC产品,云顶新耀、科伦药业、君实生物布局了Trop2 ADC产品,康诺亚/美雅珂布局了Claudin 18.2 ADC产品,恒瑞医药、荣昌生物深耕于cMET ADC产品。未来,我国ADC药物市场将呈现出百花齐放、百舸争流的繁荣景象。
风险提示:药物研发不及预期,药品降价风险,研发同质化风险



一、ADC药物发展趋势
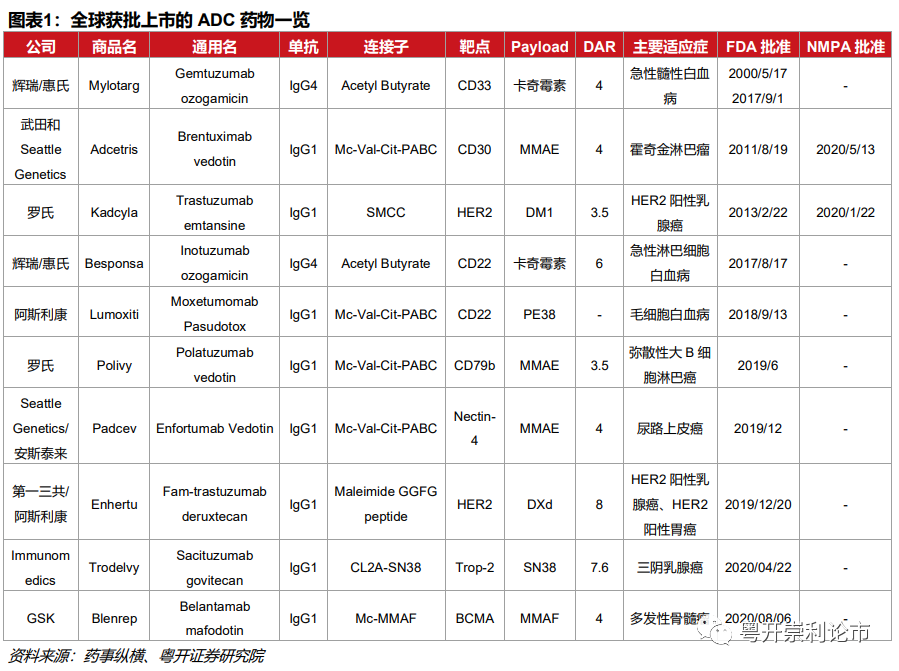
随着DS-8201革命性的成功,自2020年起,ADC药物呈现出资本竞逐的局面。截至2021年3月,全球已有十款ADC药物获批上市,包括武田/Seattle Genetics的Adcetris、罗氏的Kadcyla、辉瑞/惠氏的Besponsa、辉瑞/惠氏的Mylotarg、阿斯利康的Lumoxiti、罗氏的Pilivy、第一三共/阿斯利康的Enhertu、Immunomedics的Trodelvy、GSK的Blenrep。其中,Kadcyla和Adcetris均于2020年在国内上市。
获批上市的ADC药物共涉及8个靶点,其中CD22、CD30、CD33、CD79b、BCMA 5个靶点的适应症为血液肿瘤;HER2、Nectin-4、Trop-2的适应症为实体瘤。此外,LIV-1、TF、c-Met、Claudin等靶点正逐步从幕后走向台前。从研发的角度思考,尽管HER2、CD22、CD30等靶点较为成熟,研发风险较低,但竞争相对激烈,存在研发扎堆的风险。布局ADC药物的企业或应另辟蹊径,遴选“人迹罕至”的适应症加速创新,抢滩登陆,发挥先发优势。
ADC药品涉及的技术表现出高度的多样性、复杂性和演变性,差异化的设计带来全新的产品。在ADC药品的设计中,抗体、连接技术和细胞毒药物是三大关键考量因素。对于相关概念及HER2 ADC产品的介绍,可参考《HER 2 ADC渐入收获期,星辰大海,砥砺前行》一文,本文将重点介绍ADC药物中抗体、连接技术和细胞毒药物的发展趋势。
(一)抗体的发展趋势
(1)鼠源抗体、人鼠嵌合单抗——人源化抗体
从第一代ADC到第二、三代ADC药物,抗体经历了鼠源单抗、人鼠嵌合单抗到人源化抗体的转变,在第二、三代ADC药物中,人源化抗体已被广泛使用。在已上市的ADC药物中,靶向CD22的Lumoxiti采用鼠源IgG1抗体,靶向CD30的Adcetris采用人鼠嵌合单抗,其余ADC产品均使用人源单抗。大批使用鼠源单抗/人鼠嵌合单抗的ADC药物倒在临床试验,根源在于这类抗体可被人体免疫系统识别,当作异源抗体被快速清除。

(2)IgG4——IgG1
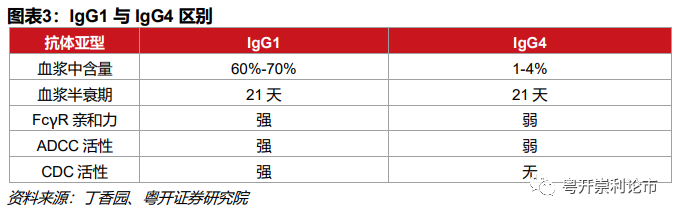
辉瑞的ADC产品普遍采用IgG4抗体,例如Mylotarg和Besponsa均使用IgG4抗体。但其他公司则更青睐IgG1抗体,这是由于IgG4不具备ADCC、CDC、ADCP作用且在低PH条件下容易聚集,而IgG1抗体不但ADCC和CDC活性较强,且拥有较高的稳定性。
但是,ADCC和CDC作用对于ADC药物发挥药效的帮助究竟几何,仍难下定论,毕竟,ADC药物主要依靠细胞毒药物发挥杀伤作用且须快速内吞。与这一机制类似的为免疫检查点抑制剂,由于免疫检查点抑制剂不依赖ADCC和CDC作用,且为了规避IgG1亚型在特殊情况下诱发细胞因子风暴的潜在风险,因此普遍采用IgG4亚型,如O药、K药、卡瑞利珠单抗。此外,尽管T药采用了IgG1亚型,但去除了糖基化,不发挥ADCC的作用。
(3)抗体亲和力提升
新一代ADC药物往往采用亲和力更强的单抗。随着抗体亲和力的提升,抗体与低表达靶点的结合能力更强,所须的药物剂量相对更低,产生的不良反应越少。抗体亲和力通常可用EC50或IC50表示,EC50和IC50的值越小,代表抗体亲和力越高。
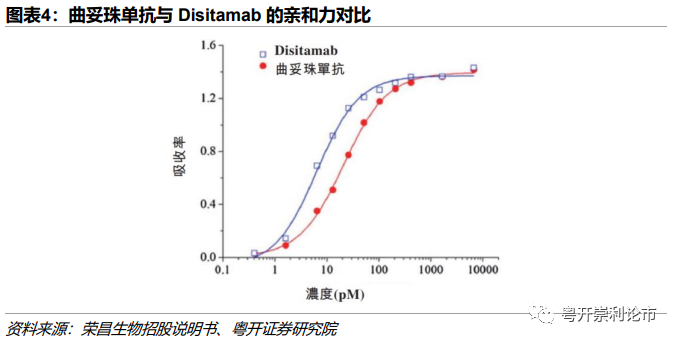
对于HER2 ADC药物,绝大多数ADC药物均使用曲妥珠单抗,荣昌生物另辟蹊径,在RC48中采用Disitamab新型抗体。Disitamab的亲和力显著高于曲妥珠单抗,与曲妥珠单抗20.1pM的EC50相比,Disitamab的EC50仅6.4pM。凭借更强的亲和力,RC48在HER2低表达的乳腺癌和胃癌上表现同样优异。
(二)Linker的发展趋势
(1)不可剪切——可剪切
可剪切的ADC药物正在成为主流。在酸或溶酶体蛋白酶的作用下,Linker在靶细胞中被切割释放出小分子毒素,小分子毒素可穿透细胞膜进入相邻肿瘤细胞,发挥旁观者效应(Bystandereffect)。
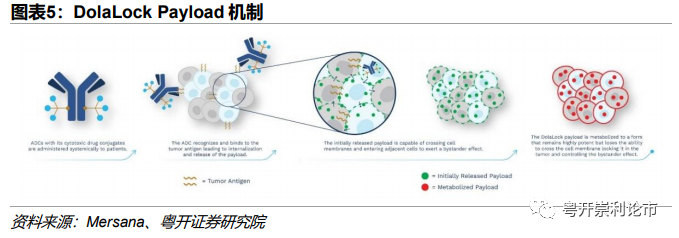
但旁观者效应是一把“双刃剑”。当相邻细胞为癌细胞时,Bystander effect可发挥积极的作用,即实现旁杀伤;但是当相邻细胞为正常细胞时,Bystander effect则会“误杀”。针对这一问题,Mersana公司研发了DolaLock Payload,该payload为Auristatin类衍生物。携带DolaLock Payload的ADC药物进入靶细胞后,ADC在特定条件下裂解,Payload发挥旁观者效应。但DolaLock Payload的旁观者效应仅可持续一段时间,随后Payload代谢为一种依旧高效、但无法跨膜的形式,将Payload“锁”在原靶细胞中,进而有效控制旁观者效应。
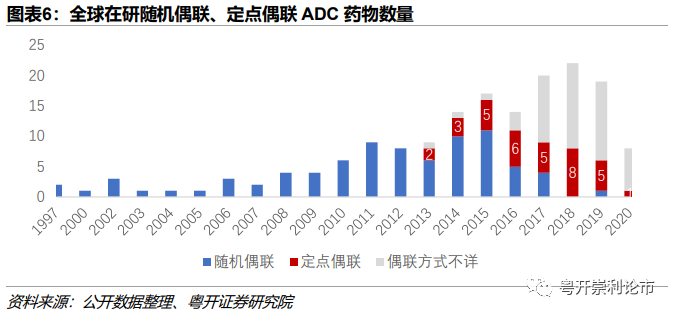
(2)随机偶联(赖氨酸——半胱氨酸)——定点偶联
随机偶联主要包括赖氨酸随机偶联与半胱氨酸随机偶联,就二者比较,半胱氨酸(-SH)的偶联方式较赖氨酸(-NH2)的偶联方式异质性更小,附着部位更容易预测。原因在于IgG仅含有4个链间二硫键,仅可产生8个有效负荷供偶联,远远低于抗体上存在的80-100个赖氨酸偶联位点。而赖氨酸偶联还会产生大量的裸抗,Mylotarg裸抗的概率高达50%,导致其一度沦为昂贵且高副作用的“单抗”药物。
新一代的ADC药物普遍采用定点偶联的连接方式,如新码生物的ARX788使用非天然氨基酸定点偶联技术,科伦药业的A166采用赖氨酸定点定量偶联技术等。但定点偶联是否真的优于随机偶联,目前仍难下定论,毕竟目前尚无定点偶联的ADC产品获批上市,而Seattle Genetics的定点偶联产品已全部宣告失败。此外,定点偶联往往产生DAR在2-4区间的ADC药物,低DAR+高毒性Payload一度被认为是最优ADC药物的设计模式,但这一概念随着DS-8201(Enhertu)和Trodelvy的上市而备受质疑。
(3)疏水性——亲水性
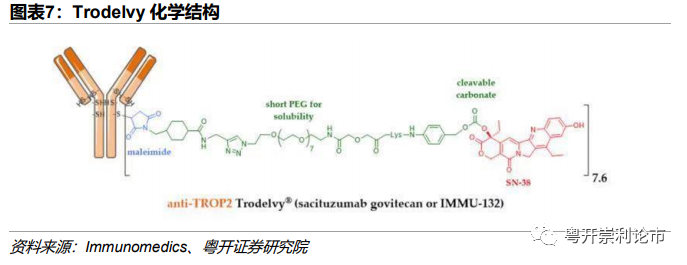
ADC药物的疏水性问题可能导致癌细胞上调MDR1的表达,产生过量P-糖蛋白,将ADC药物泵出细胞外,减低了细胞内的药物浓度并使细胞产生多重耐药性(MDR)。由于P-糖蛋白作用的常为疏水性分子,因此可通过改善药物在水中的溶解度削弱MDR效果。在药物的设计中,可使用聚乙二醇(PEG)连接物调整药物水油分布系数。Immunomedics研发的Trodelvy便运用了较短的聚乙二醇化单元连接体结构,改善了药物在水中的溶解度,可能有助于对抗ADC耐药。
(4)偶联组合
偶联组合基于各公司的技术平台。如辉瑞采用Acetyl Butyrate+卡奇霉素的组合、基因泰克采用SMCC+DM1的组合、Seattle Genetics采用Mc-Val-Cit-PABC+MMAE的组合、第一三共采用马来酰亚胺-GGFG+Dxd的组合。不同公司的偶联组合各有千秋,并不存在完美的组合,如Mc-Val-Cit-PABC中的马来酰亚胺可在血浆中发生逆向Michael加成反应,导致去共轭,或诱发脱靶毒性。
(三)Payload的发展趋势
(1)微管蛋白抑制剂——DNA抑制剂
ADC药物的Payload主要包括微管蛋白抑制剂和DNA抑制剂。在微管蛋白抑制剂中,美登素类和Auristatin类使用最广泛。在二者的选择上,由于Auristatin类的毒性较美登素类更强,越来越多的ADC药物倾向于使用MMAE或MMAF。通过梳理已上市的ADC产品,仅Kadcyla使用DM1作为Payload,Adcetris、Polivy和Padcev均使用MMAE,Blenrep使用MMAF。
就MMAE和MMAF比较,由于MMAF可产生具有带负电荷的羧基末端苯丙氨酸残基的代谢物,因此无法像MMAE跨膜发挥旁观者效应。MMAF的优势在于药物的最大耐受量较高,不需要可裂解Linker也可高效地发挥抗肿瘤作用。
除了广泛运用的微管蛋白抑制剂,DNA抑制剂正在崭露头角,Enhertu和Trodelvy分别使用DXd和SN-38作为payload。DXd和SN-38均是DNA拓扑异构酶Ⅰ抑制剂,前者的活性是伊立替康的10倍,干扰DNA复制、重组和基因表达;SN-38是伊立替康的活性代谢物。与微管蛋白抑制剂相比,DNA抑制剂具有诸多优势:
第一,细胞内微管蛋白抑制剂的靶标数量远超DNA抑制剂的靶标数量,在ADC携带相同数量弹头进入细胞的情况下,DNA抑制剂能发挥更好的杀伤作用;
第二,微管蛋白抑制剂半衰期较长,DNA抑制剂半衰期短,不易在体内发生蓄积;
第三,微管蛋白抑制剂抗癌谱较窄,对于结直肠及大部分胃肠道肿瘤活性较低,可能不适用于不敏感肿瘤的治疗,Kadcyla对晚期胃癌已宣布无效。
从这些特性分析,我们认为,DNA抑制剂未来有望替代微管蛋白抑制剂。

(2)低DAR——高DAR
在传统ADC药物的设计中,由于化学接头的局限性,不得不使用高毒性的Payload并采用DAR为3~4的设计。但随着DS-8201和Trodelvy的巨大成功,科学家将目光投向高DAR的设计中,前者采用了DAR为8的设计,后者的DAR高达7.4。通过高DAR+中等毒性Payload的组合,不但可以更好地发挥抗肿瘤作用,而且脱靶毒性有望降低。
目前,科学家们正将目光投向更高DAR的药物。Mersana公司的Fleximer平台可设计出DAR达10-12的ADC药物。Fleximer是一种可生物降解的、高度生物相容的、水溶性的聚合物,一端通过可剪切连接子连接细胞毒药物,一端通过不可剪切连接子连接单抗,提升ADC药物的DAR值,改善效应分子的运输效率。
但是,值得注意的是,高DAR的设计或牺牲了ADC药物的稳定性和均一性,可能引起抗体的集聚,影响ADC药物的药效。
二、CD30-ADC
CD30是肿瘤坏死因子(TNF)受体超家族成员之一。健康个体中,CD30低表达于正常激活的T细胞和B细胞表面,在正常细胞中不表达。在霍奇金淋巴瘤和和间变大细胞淋巴瘤表面,CD30常处于高表达。此外,研发发现CD30与细胞的增殖和死亡密切相关,特别是其胞内结构部分可与TNFR相关因子家族的多个成员相互作用,不仅能够通过JNK和p38途径诱导细胞凋亡,还可以依赖NF-κB途径介导细胞活化。
CD30的以上特征符合理想靶抗原的特点:在靶细胞表面高度一致性表达,在正常组织低表达,差异化的表达模式可减少脱靶(肿瘤外)毒性;此外,靶抗原脱落概率极低,防止游离抗原与循环中的mAb结合造成的失效。

(一)Adcetris :血液肿瘤领域最成功的ADC药物
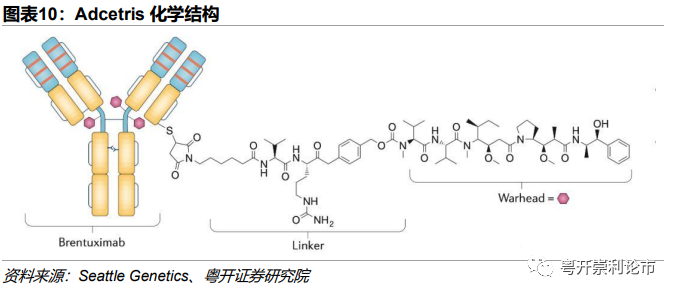
Brentuximab Vedotin(维布妥昔单抗)商品名为Adcetris,是一种靶向CD30的抗体偶联药物(ADC)。Adcetris由3个组分构成:(1)靶向CD30的人鼠嵌合单抗Brentuximab;(2)瓜氨酸-缬氨酸随机偶联的二肽连接子;(3)小分子药物MMAE(Auristatin E),DAR=4。
FDA批准:Adcetris由西雅图遗传公司(Seattle Genetics)研制,2009年,西雅图遗传与武田制药达成协议,后者获得Adcetris除美国和加拿大以外国家的商业化权力。2011年,Adcetris获FDA批准上市,目前已获批六个适应症。
NMPA批准:2020年5月,Adcetris(安适利)获NMPA批准上市,用于治疗复发或难治性系统性间变性大细胞淋巴瘤或CD30阳性霍奇金淋巴瘤成人患者。

作用机制:当Adcetris与肿瘤细胞膜CD30受体结合后,Adcetris主要通过网格蛋白介导的内吞作用内化进入溶酶体,随后二肽被蛋白酶水解断裂,最终MMAE与细胞质的微管蛋白结合从而阻滞细胞周期的G2/M期,使细胞死亡。由于采用了可裂解的Linker,MMAE可透过细胞膜作用于周围细胞,对CD30非阳性肿瘤细胞产生细胞毒效应。

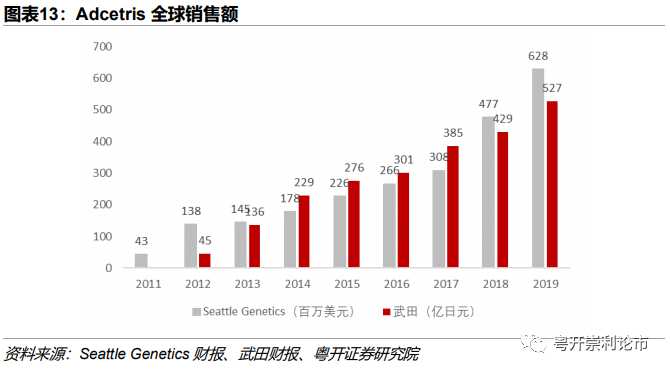
销售数据:自2011年上市以来,Adcetris市场一路高歌猛进。2019年,Adcetris美国和加拿大市场销售额达6.28亿美元,其他市场销售额达527亿日元。据Clarivate预测,Adcetris2020年全球市场将达11.7亿美元,2024年将突破20亿美元。
系统性间变性大细胞淋巴瘤(sALCL)
实验设计:Adcetris系统性间变性大细胞淋巴瘤的获批基于SG035-0004试验。SG035-0004研究是一项关键性、Ⅱ期、单臂、开放标签的多中心临床试验,旨在评估Adcetris单药治疗复发或难治性sALCL的疗效和安全性。研究纳入58例年龄≥12岁,可测量病灶≥1.5 cm和氟脱氧葡萄糖摄取、ECOG体能状态评分为0或1、既往接受≥1种治疗方案、CD30阳性复发或难治性sALCL患者,给予维布妥昔单抗治疗最多16个疗程。主要终点是由IRF评估的ORR。
实验结果
疗效数据:58例患者中97%的复发或难治性sALCL患者实现肿瘤缩小,客观缓解率(ORR)达86%,完全缓解(CR)率达57%,部分缓解(PR)率达29%。5年长期随访数据显示,总人群5年生存率(OS)达60%,5年无进展生存率(PFS)达39%,中位PFS为20个月。5年38例(66%)获得完全缓解(CR),完全缓解率达79%,5年PFS率达57%。
安全性数据:周围神经病变(PN)发生率为57%(33/58例)。PN可控且可逆,在末次随访时,91%PN患者的症状完全消退(67%)或改善(24%)。Adcetris于2012年收到FDA的进行性多灶性白质脑病的黑框警告。

CD30阳性霍奇金淋巴瘤
实验设计:CD30阳性霍奇金淋巴瘤适应症的获批基于SG035-0003。
SG035-0003是一项关键性、Ⅱ期、单臂、开放标签、多中心临床试验,旨在评估Adcetris单药用于自体造血干细胞移植后复发或难治性CD30阳性HL患者的疗效和安全性。研究共纳入102例患者,为年龄≥12岁、可测量病灶≥1.5 cm、ECOG体力状态评分为0或1、既往接受大剂量化疗和自体造血干细胞移植(ASCT)、CD30阳性的复发或难治性HL患者,给予维布妥昔单抗治疗最多16个疗程。主要终点是由独立审查机构(IRF)评估的客观缓解率(ORR)。
实验结果
疗效数据:在SG035-0003研究中,94%的复发或难治性cHL的患者实现肿瘤缩小,整体人群ORR为75%,CR率为34%,疾病控制率(DCR)为96%。获得ORR患者的中位DOR为6.7个月,获得CR患者的中位DOR接近2年。5年长期随访数据显示,中位总生存期(OS)和PFS分别为40.5个月和9.3个月,估计的5年OS率和PFS率分别为41%和22%,获得CR患者5年OS率的PFS率分别为64%和52%。
安全性数据:不良反应总体可控,56例(55%)患者出现PN,但症状恢复比例较高,73%的患者可以完全消退(41/56例),14%改善(8/56例)。
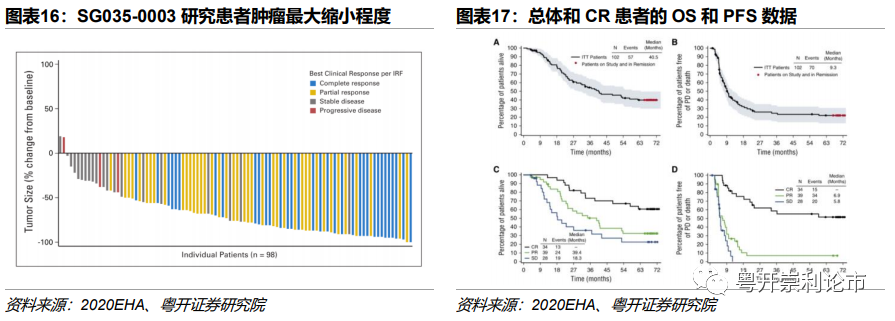
非干细胞移植候选复发或难治性cHL患者
实验设计:C2507研究为一项4期单臂临床试验,研究共纳入60例既往至少接受一次化疗方案且开始维布妥昔单抗治疗时不适合进行干细胞移植(SCT)或多药治疗的复发或难治性cHL患者。中位治疗7个周期,在后续治疗中,28例(47%)的患者在接受中位数为7个周期的维布妥昔单抗治疗后继续接受SCT,而32例(53%)患者在接受中位7周期维布妥昔单抗治疗后未接受后续SCT。
实验结果
疗效数据:根据IFR评估,意向性治疗人群(ITT)的ORR为50%。在30例CR(7例)或PR(23例)患者中,至缓解的中位时间为6周,至最佳总体缓解的中位时间为11周。
接受>1次既往癌症相关治疗的患者,ORR为51%,6例(12%)患者报告的CR的最佳总体缓解,19例(39%)患者报告PR的最佳总体缓解。接受一次既往癌症相关治疗的患者的ORR为45%,1例(9%)报告CR,4例(36%)报告PR。
(二)国内CD30ADC:复旦张江/交联药物独家布局
国内针对CD30进行ADC布局的企业数量较少,目前处于临床阶段的仅有复旦张江/交联药物的F0002-ADC。F0002由三个部分组成:人鼠嵌合抗CD30单克隆抗体、硫醚连接子(MCC)和DM1。F002于2016年启动立项,适应症为复发/难治性CD30阳性血液肿瘤,2018年1月完成临床前研究,2019年3月14日启动Ⅰ期临床,目前处于招募中状态。
三、CD22-ADC
CD22属于免疫球蛋白超家族和SIGLEC(唾液酸结合Ig样凝集素)家族,属于B细胞表面抑制性辅助受体之一,与B细胞的发展、分化和功能密切相关。研究发现,CD22在大多数B细胞恶行肿瘤细胞表面均有表达,包括急性淋巴细胞白血病(ALL)、非霍奇金淋巴瘤(NHL)和慢性淋巴细胞白血病(CLL)。Immunomedics是最早研究CD22的企业之一,早期以研发CD22单抗为主,但由于CD22容易内吞导致杀伤癌细胞作用较弱,因此对CD22研发的重点由单抗逐步转向ADC药物。目前,Besponsa和Lumoxiti均获得FDA批准上市。针对CD22 ADC药物,国内尚无企业进入临床阶段。
(一)Besponsa:FDA批准的首款靶向CD22的ADC药物
Inotuzumabozogamicin(伊珠单抗奥唑米星)商品名为Besponsa,是辉瑞公司与合作伙伴联合开发的一款ADC药物。Adcetris由3个组分构成:(1)靶向CD22的人源IgG4单抗Inotuzumab;(2)可剪切的腙键;(3)DNA抑制剂卡奇霉素,DAR=6。
技术:Besponsa使用的ADC技术较为古老,Linker技术为第一代的可剪切的腙键连接,腙键依靠酸水解,因此在血液中存在的时间有限。鉴于此,腙键局限于血液肿瘤,无法运用于实体瘤,因为ADC药物可在30分钟内到达血液肿瘤组织。首个上市的、靶向CD33的ADC药物Mylotary同样采用腙键和卡奇霉素,适应症为急性髓性白血病(AML),但因安全性太差撤市,之后降低了药物剂量再次上市。
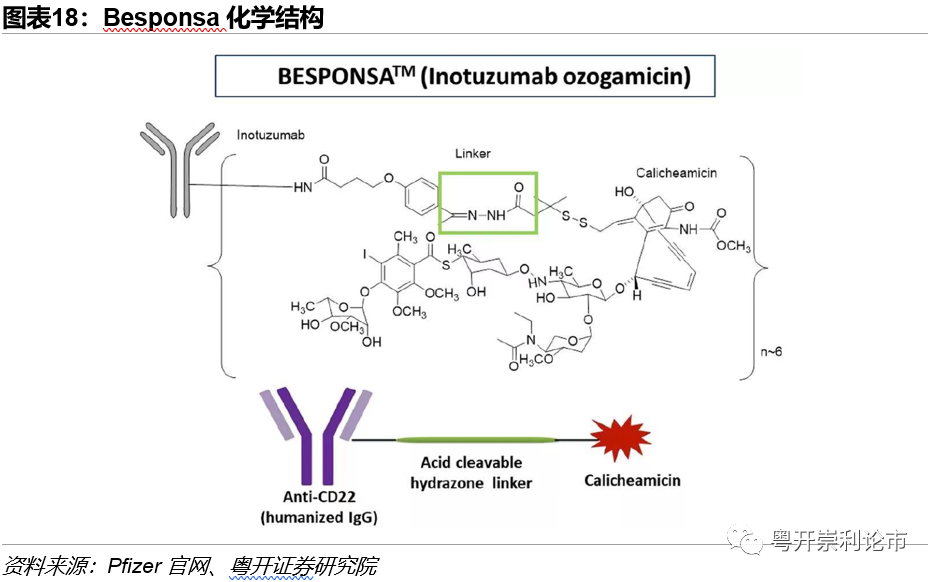
FDA批准:2017年8月,美国FDA加速批准Besponsa上市,适应症为成人复发或难治性B细胞前体急性淋巴性白血病(ALL),Besponsa为FDA批准的首款靶向CD22的ADC药物。
NMPA递交上市申请:2020年1月,辉瑞向NMPA递交了Besponsa的上市申请,适应症为复发性或难治性CD22阳性的急性淋巴细胞白血病。2020年3月9日,Besponsa纳入优先审评范围,这将进一步加速Besponsa在中国的上市进程。
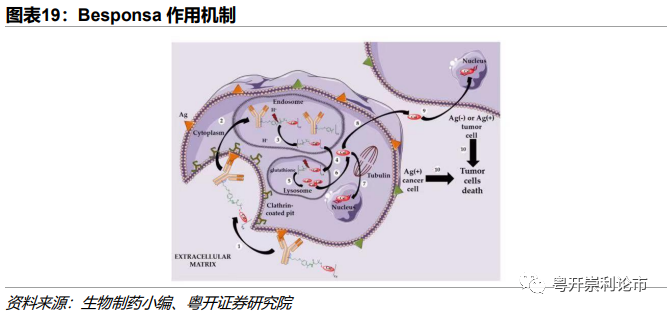
作用机制:CD22抗原在B细胞表面普遍存在,Besponsa可通过靶向癌细胞,与其表面的CD22抗原结合,内吞后进入癌细胞溶酶体,ADC药物裂解后,卡奇霉素从偶联物上水解游离,进入细胞核,与DNA小沟结合。在卡奇霉素的作用下,DNA双螺旋链断裂,癌细胞死亡。
销售数据:辉瑞并未公布Besponsa的销售数据,销售收入可能不足1亿美元。这源于ALL为相对罕见的疾病,美国成人ALL年发病人数约3000人,死亡人数约1000人。此外,Besponsa尽管CR和PFS明显优于化疗,但OS并未明显改善。
急性淋巴瘤白血病(ALL)
实验设计:Besponsa的ALL适应症的获批基于Ⅲ期INO-VATE ALL试验结果,试验为一项随机、开放标签、国际、多中心研究,纳入326例成人复发或难治性B细胞ALL患者,患者被随机分为两组,分别接受Besponsa或标准化疗。主要终点是完全缓解率(CR)。
实验结果
疗效数据:试验结果显示,Besponsa组中患者的完全缓解率(CR)为81%(95% CI:72%-88%),化疗组的CR仅为29%(95% CI:21%-39%)。在所有CR患者中,接受Besponsa治疗的患者其最小病灶残留(MRD)的阴性率为78%(95% CI:68%-87%),化疗组的这一数字为28%(95% CI:14%-47%)。此外,Besponsa组患者的中位数总生存期(OS)为7.7个月,化疗组数据为6.2个月,OS并未有明显改善。
安全性数据:Besponsa携有严重肝损伤的黑框警告。常见的不良反应为血小板降低、白细胞减少、中性粒细胞减少、感染、贫血、疲乏、肝损伤(ALT和AST升高)。FDA建议,孕妇和哺乳期女性慎用Besponsa,该药可能对胎儿和新生儿发育存在不良影响。
(二)Lumoxiti:开启毛细胞白血病治疗新纪元
MoxetumomabPasudotox商品名为Lumoxiti,是阿斯利康研发的一款ADC药物。Adcetris由靶向CD22的鼠源IgG1单抗Moxetumomab和截短的假单胞菌外毒素PE38组成。
Lumoxiti适应症为毛细胞白血病。毛细胞白血病(HCL)是一种罕见的、无法治愈的、进展缓慢的淋巴增值性慢性白血病,以贫血、出血、脾脏肿大以及外周血及骨髓出现大量边缘不整齐呈伪足状或纤毛样突出的白细胞为特征。毛细胞白血病可导致骨髓生成过多的异常B细胞,从而阻止健康血细胞的产生。
技术:Lumoxiti是目前唯一获批的免疫毒素ADC药物。免疫毒素是用于选择性破坏具有某一特异性标记细胞的一类融合蛋白,通常是由具有高度特异性的单克隆抗体与具有强大杀伤作用的毒素分子通过化学交联构建而形成。免疫毒素具有高效、低毒、高特异性等特点,但免疫毒素也存在不少问题,如对靶组织的特异性杀伤作用弱,诱发不同程度的血管渗透综合征(VLS)等。
FDA批准:2018年9月,Lumoxiti获FDA批准用于既往已接受过至少两种系统疗法(包括嘌呤核苷类似物)治疗失败的复发性或难治性毛细胞白血病(HCL)成人患者的治疗。Lumoxiti是20年来首个被批准的用于复发以及难治性毛细胞白血病的新疗法。
EMA批准:2021年2月13日,Lumoxiti获欧盟委员会批准上市,适应症与FDA相同。
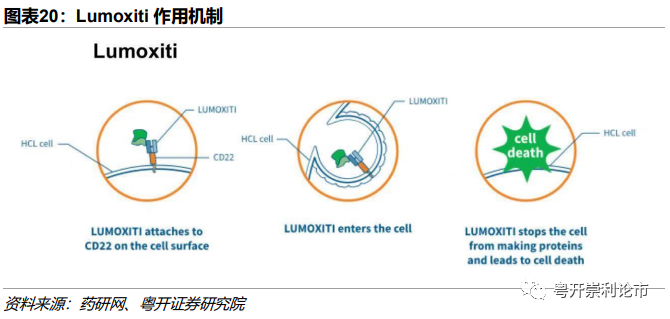
作用机制:Lumoxiti与B细胞CD22靶点结合后,药物内吞,细胞毒素PE38可抑制蛋白质的合成,诱导靶细胞凋亡。
毛细胞白血病(HCL)
实验设计:Lumoxiti在美国和欧盟的获批基于关键性Ⅲ期(Study 1053)临床研究的数据,该研究为一项单臂、多中心研究,在80例既往接受了至少2种方案的复发性或难治性HCL成人患者种开展,评估了Lumoxiti单药治疗的疗效和安全性。
实验结果
疗效数据:试验结果显示,Lumoxiti单药治疗的总缓解率(ORR)为75%,持久的完全缓解率(CR)为36%(29/80),定义为血液学缓解至少维持180天的完全缓解(CR)。有81%的CR患者经理了微小残留病灶的根除,即MRD阴性状态。此外,病情获得CR的患者在5年后维持CR的概率为61%。
安全性数据:Lumoxiti的常见3级或4级不良反应(至少≥5%)包括高血压、发热性中性粒细胞减少症、毛细血管渗透综合征(CLS)、溶血性尿毒综合征(HUS)等,一般副作用包括头晕、呼吸困难、呕吐、发烧、疲惫等。Lumoxiti不推荐用于存在严重肾功能损害的患者。
四、Trop2 ADC
Trop2是由TACSTD 2基因编码表达的细胞表面糖蛋白,又名肿瘤相关钙离子信号转导子2(TACSTD2)。研究表明,TROP2在人体健康组织中有不同水平的表达,表达水平最高的是乳腺、肾脏以及胰脏的上皮组织。在肿瘤中,TROP2的表达水平会出现显著升高,包括乳腺癌、肺癌、胃癌、结肠直肠癌、胰腺癌、前列腺癌、宫颈癌等,科学家认为TROP2的高表达在肿瘤细胞的生长过程中起到关键作用,还与更侵袭性的疾病和预后不良相关。
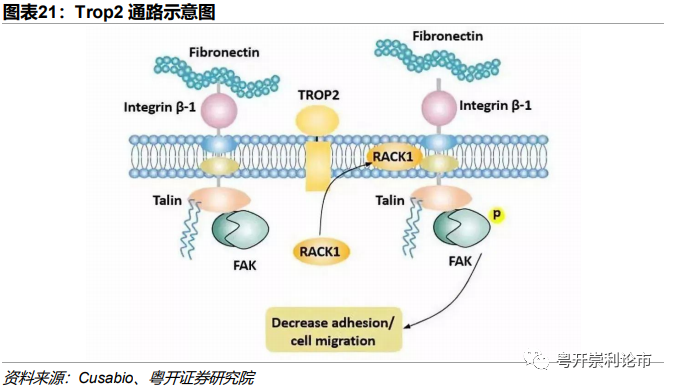
资料来源:Cusabio、粤开证券研究院 |
三阴乳腺癌(Triple Negative BreastCancer, TNBC)为一种预后较差的侵袭性癌症,在乳腺癌中占比约15%~20%。由于雌激素受体、孕激素受体和人表皮生长因子受体-2(HER2)三个重要基因表达均为阴性,导致内分泌治疗及抗HER2靶向治疗对其疗效甚微,除了传统化疗以外,其他治疗方案十分有限。在三阴乳腺癌细胞中,由于90%以上的细胞表面糖蛋白均会出现TROP2的高表达,因此,科学家将目光投入Trop2药物的研发,以期治疗TNBC。
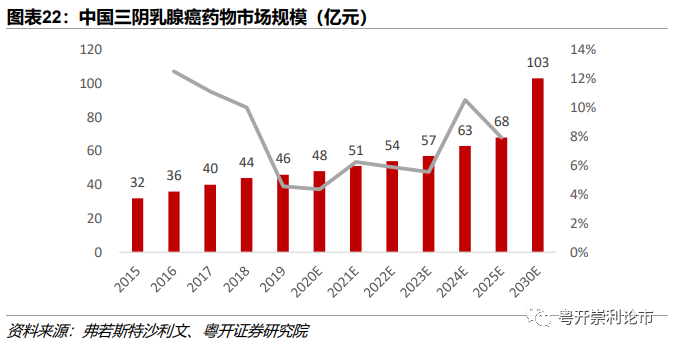
2024年我国Trop2ADC药物市场规模有望达7.56-9.45亿元。我国TNBC发病人数自2015年至2019年以1.8%的年复合增长率由4.6万人增加至4.9万人。预计到2024年新发人数将达到5.3万人,到2030年新发人数达5.6万人。从中国TNBC药物市场规模,2015年我国TNBC市场规模达32亿元人民币,2019年增长至45亿元人民币,2024年我国TNBC市场规模有望达63亿元,2030年达到103亿元。我们预计,Trop2 ADC药物上市后,由于价格远高于PD-1/L1等免疫疗法,市场渗透率预计达12%-15%,2024年市场规模有望达7.56-9.45亿元。
(一)Trodelvy:首个治疗mTNBC的ADC药物
SacituzumabGovitecan商品名为Trodelvy,由Immunomedics(已于2020年9月被吉利德以210亿美元收购)研发,是目前全球唯一一款获批上市的靶向Trop2的ADC药物,适应症为三阴乳腺癌(TNBC)。Trodelvy由3个组分构成:(1)靶向Trop2的人源单抗Sacituzumab;(2)马来酰亚胺连接体;(3)拓扑异构酶Ⅰ抑制剂SN-38(伊立替康的活性代谢物),DAR=7.4。
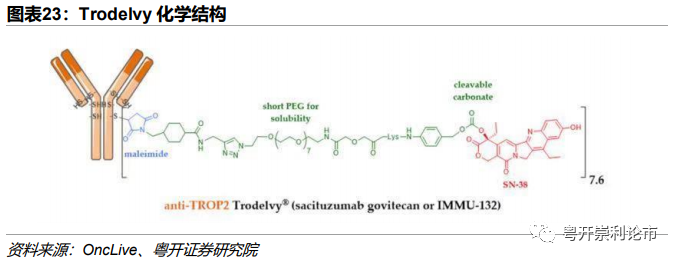
技术:1)Trodelvy采用中等毒性的拓扑异构酶抑制剂SN-38,有效规避了微管抑制剂抗肿瘤谱狭窄等问题,进一步将适应症拓宽至三阴乳腺癌。通过中等毒素和高DAR的设计,药物的抗肿瘤效力进一步提升;2)较短的聚乙二醇化单元连接体结构克服了药物的疏水性问题,改善了药物在水中的溶解度,可能有助于对抗ADC耐药;3)可裂解Linker,SN-38进入旁细胞发挥Bystander Effect。
FDA批准:2020年4月,Trodelvy获得美国FDA加速批准上市,用于先前已接受过至少两种疗法治疗转移性疾病的转移性三阴性乳腺癌成人患者。Trodelvy是FDA批准的首个专门治疗复发或难治性mTNBC的ADC药物。
NMPA递交临床申请:2019年4月,云顶新耀获得Trodelvy在大中华区、韩国及部分东南亚国家的独家权益,涉及金额高达8.35亿美元。2020年5月,Trodelvy获NMPA批准开展针对mTNBC的Ⅰ期临床。2020年12月9日,云顶新耀公告Trodelvy已完成用于转移性乳腺癌(mBC)亚洲3期临床试验首例患者给药。
新加坡递交上市申请:2021年1月6日,云顶新耀已向新加坡卫生科学局递交Trodelvy用于治疗接受过至少两线既往治疗的转移性三阴乳腺癌患者(mTNBC)的新药上市申请。
(1)转移性三阴乳腺癌(mTNBC)ASCENT Ⅱ期
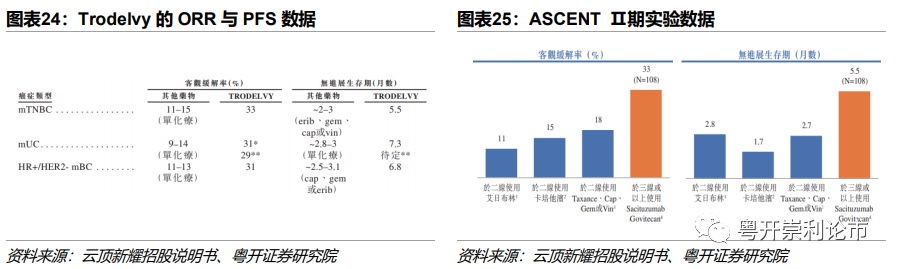
实验设计:Trodelvy的mTNBC适应症的获批上市基于ASCENT研究,研究纳入108名既往至少接受过2种标准化放疗进展复发的转移性三阴性乳腺癌患者,以1:1比例随机分配到Trodelvy或TPC化疗组。Trodelvy治疗组患者在重复的21天周期的第1、8天接受10mg/kg的剂量作为既定剂量方案展开研究。患者每8周进行一次肿瘤成像,主要终点为客观缓解率(ORR)与缓解持续时间(DOR)。
实验结果
疗效结果:在108名曾接受至少二线治疗转移性TNBC患者中,客观缓解率(ORR)达33.3%,其中完全缓解率(CR)达2.8%,PR达30.6%,远高于化疗组的疗效。在对治疗有反应的39例患者中,中位缓解持续时间(mDOR)为7.7个月,中位无进展生存期(mPFS)为5.5个月。
安全性数据:治疗常见的副作用为恶心,中性粒细胞减少,腹泻,疲劳,贫血,呕吐,脱发,便秘,食欲下降,皮疹和腹痛。
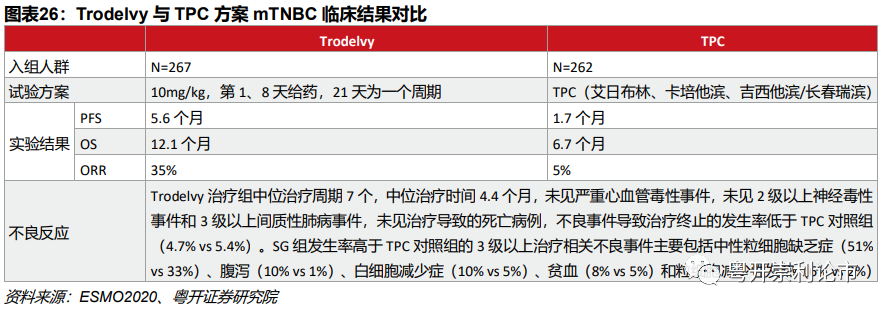
(2)转移性三阴乳腺癌(mTNBC)ASCENTⅢ期
实验设计:ASCENT研究是一项国际多中心、开放标签、随机、平行组的Ⅲ期试验,入组了529例既往接受过至少两种化疗(包括紫杉醇)的复发难治mTNBC,分别给予Trodelvy和TPC(艾日布林、卡培他滨、吉西他滨或长春瑞滨),研究的主要终点的未发生脑转移患者的PFS,次要终点包括全部患者的PFS、OS、ORR、DoR和安全性。
实验结果
疗效数据:在主要终点方面,Trodelvy组的中位PFS为5.6个月(4.3~6.3),TPC组为1.7个月。在次要终点方面,Trodelvy相比TPC组显著改善了患者OS(12.1VS6.7个月),ORR(35%VS 5%)
安全性数据:Trodelvy的药品标签中带有黑框警告,提示有严重中性粒细胞减少和腹泻的风险。Trodelvy治疗组不良反应发生率低于TPC对照组(4.7%VS 5.4%),Trodelvy组发生率高于TPC对照组的3级以上TRAE主要包括中性粒细胞缺乏症(51%VS 33%)、腹泻(10%VS 1%)、白细胞减少症(10%VS 5%)、贫血(8%VS 5%)和粒细胞减少性发热(6% VS 2%)。
(3)尿路上皮癌
实验设计:2020年9月,在ESMO2020虚拟大会上,Immunomedics公布Trodelvy在治疗转移性尿路上皮癌(mUC)的关键性Ⅱ期临床试验的结果。
实验结果
疗效数据:Trodelvy达到31%的ORR,CR达5%,mPFS达5.4个月,mOS达10.5个月。对比使用含铂治疗和免疫检查点抑制剂的患者PFS仅为2-3个月,优势较为明显。
结论:尿路上皮癌适应症有望于2021年获批上市。尽管与含铂与免疫检查点抑制剂相比,Trodelvy优势明显,但与Padcev数据相比,数据难言亮眼。我们认为Padcev有望在尿路上皮癌领域树立新的标准。目前Gilead也在探索Trodelvy与Keytruda联用的试验。
(二)DatopotamabDeruxtecan(DS-1062):晚期NSCLC的潜在用药,已启动Ⅲ期临床
DS-1062是由第一三共和阿斯利康联合开发靶向Trop2的ADC药物,目前DS-1062对比多西他赛治疗经治的晚期或转移性NSCLCⅢ期临床已经开始招募患者。DS-1062由3个组分构成:(1)靶向Trop2的人源IgG1单抗Datopotamab;(2)可裂解半胱氨酸Linker;(3)拓扑异构酶Ⅰ抑制剂DXd,DAR=4。
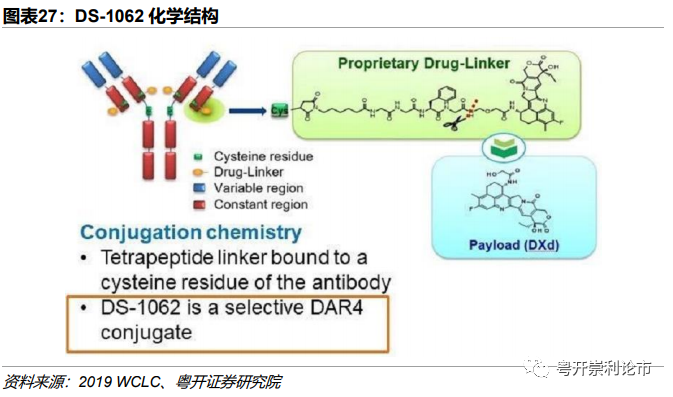
技术:1)DXd为一种创新DNA拓扑异构酶Ⅰ抑制剂,活性为伊立替康(SN-38)的十倍,可干扰DNA复制、重组和基因表达。与微管蛋白抑制剂相比,DXd为更有效和更有潜力的化疗药物;2)半胱氨酸的偶联方式较赖氨酸异质性更小,附着部位更容易预测;3)可剪切linker,发挥旁观者效应。
里程碑费用:阿斯利康和第一三共将平摊全球范围内(除日本)与DS-1062相关的开发和商业化费用以及利润,第一三共将承担在日本的费用并向阿斯利康支付个位数的版税。根据协议,阿斯利康将分期向第一三共支付10亿美元的预付款,即协议完成时支付3.5亿美元,从协议生效之日算起,12个月后支付3.25亿美元,24个月后支付3.25亿美元。在该药物成功获批上市时,阿斯利康将额外支付高达10亿美元监管里程碑款项,另外还有高达40亿美元的销售相关里程碑款项,总交易额高达60亿美元。

非小细胞肺癌(NSCLC)
在2019年世界肺癌大会上(WCLC19)上,研究者公布了DS-1062用于治疗晚期NSCLC的首次人体实验数据。
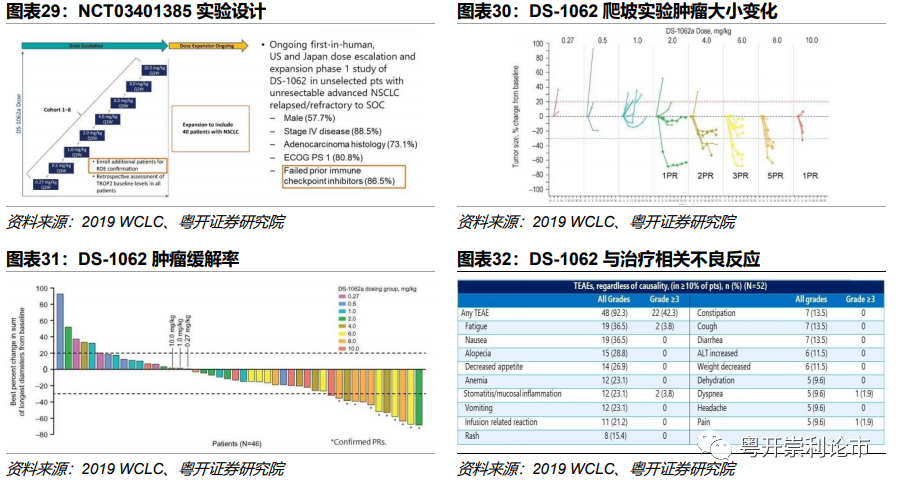
实验设计:NCT03401385为首次人体实验,实验入组了40名晚期NSCLC患者,其中男性占57.5%。所有患者均接受过包括EGFR、ALK抑制剂和免疫检查点抑制剂在内的反复治疗,平均经过3.5种方案的治疗,其中免疫检查点抑制剂失败的占比达86.5%。爬坡药物剂量范围达0.27~10mg/kg,Q3W。本次研究目的是为了探索DS-1062的安全性和耐受性并确定最大耐受剂量(MTD)和扩展剂量(RDE)。
实验结果
疗效结果:实验结果显示,12人获得部分缓解(PR),其中10人已得到确认,2例有待进一步疗效确认。对于非小细胞肺癌,其中64%的非小细胞腺癌和75%非小细胞鳞癌中存在Trop2高表达,对于Trop2高表达的患者有更高的部分缓解率(PR)。
从剂量的角度,DS-1062对于多线治疗后的耐药的非小细胞肺癌有不错的疗效,至少40%的患者肿瘤缩小30%甚至更多,而且有仅80%的患者肿瘤控制良好。专家认为, DS-1062的MTD和RDE为8mg/kg。
安全性结果:可评估安全性的52名患者的初步数据显示,DS-1062在中位治疗暴露时间为10.6周,最常见的不良反应包括疲劳(36.5%)和恶心(36.5%),三级以上的不良反应主要为疲劳(2%)、口腔黏膜炎(2%)、呼吸困难(1%)和疼痛(1%)。
2020年世界肺癌大会上(WCLC20)披露了DS-1062治疗晚期NSCLC患者的Ⅰ期临床研究结果。
实验设计:研究纳入175名既往其他标准治疗失败(包括其他靶向药、PD-1/L1、传统化疗等)的晚期非小细胞肺癌患者,分为三个剂量组(4mg/kg、6mg/kg和8mg/kg)、每三周一次接受DS-1062治疗。本次研究的主要终点是探索DS-1062的MTD、安全性和耐受性,次要终点是疗效和PK。
实验结果
疗效结果:175例晚期NSCLC患者接受了DS-1062单药三种剂量水平(4mg/kg 50例,6mg/kg 45例,8mg/kg 80例)的治疗,截至2020年9月4日,DS-1062的临床疗效为:
4mg/kg剂量组:ORR 23%,DCR 73%,PFS 4.3个月;
6mg/kg剂量组:ORR 21%,DCR 67%,PFS 8.2个月;
8mg/kg剂量组:ORR 25%,DCR 80%,PFS 5.4个月。
安全性结果:DS-1062的主要不良反应包括恶心、口腔黏膜炎、脱发、乏力等。其中14例(8%)患者发生间质性肺炎,其中4mg/kg组1例(3级)、6mg/kg组1例(2级)、8mg/kg组12例(8例1-2级,1例3级,3例5级)。
结论:从安全性的角度考虑,4mg/kg和6mg/kg较8mg/kg安全性和耐受性更好, 6mg/kg的剂量被确定为DS-1062的MTD。目前,DS-1062对比多西他赛治疗经治的晚期或转移性NSCLC的三期临床已经处于患者招募。

(三)国内Trop2ADC布局
国内共三家企业着手Trop2 ADC的研发,分别是云顶新耀的IMMU-132、科伦药业的SKB264和君实生物/多禧生物的JS108。截至2021年3月16日,云顶新耀的IMMU-132研发居前,已处于临床Ⅱ期;科伦药业的SKB264处于Ⅰ/Ⅱ期;君实生物的JS108处于临床Ⅰ期。
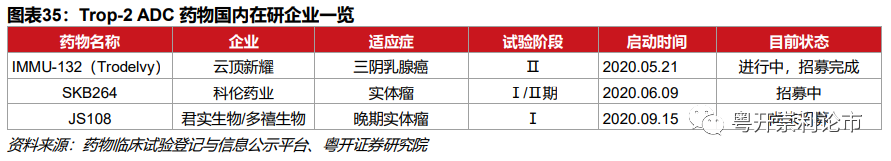
云顶新耀:2019年4月,云顶新耀获得Trodelvy在大中华区、韩国及部分东南亚国家的独家权益,涉及金额高达8.35亿美元。2020年5月,Trodelvy获NMPA批准开展针对mTNBC的Ⅰ期临床。2020年12月9日,云顶新耀公告Trodelvy已完成用于转移性乳腺癌(mBC)亚洲3期临床试验首例患者给药。
科伦药业:SKB264是科伦博泰自主研发的TROP-2 ADC药物,含有三个主要组成部分:1)重组抗Trop2人源化单克隆抗体;2)含2-(甲基磺酰基)嘧啶接头的连接子;3)拓扑异构酶抑制剂KL610023。2020年4月9日,科伦博泰的SKB264获得国家药监局临床试验通知书。
目前,SKB264处于Ⅰ/Ⅱ期临床,其中Ⅰ期的主要目的是确定最大耐受剂量(MTD)和Ⅱ期推荐剂量(RP2D);Ⅱ期的主要目的是评估SKB264单药治疗的客观缓解率(ORR),由于中美已完成三个剂量组爬坡,Ⅱ期拟先行在4mg/kg剂量下启动针对三阴乳腺癌、尿路上皮癌和卵巢上皮癌的试验。
君实生物/多禧生物:JS108为注射用重组人源化抗Trop2单抗-Tub196偶联剂,用于治疗三阴乳腺癌、肺癌和胰腺癌。2019年12月,君实生物通过独占许可授权方式从多禧生物获得JS108的许可使用权,双方约定3000万元的首付款、不超过2.7亿的研发和上市里程碑费用,以及年销售收入6%-10%的收益分成。2020年7月21日,JS108获NMPA核准签发的《药物临床试验批准通知书》,用于晚期实体恶性肿瘤的临床试验。2020年11月25日,上海君实生物的JS108的1期临床研究已完成首例患者给药。
五、其他热门ADC靶点
(一)EpCAMADC:齐鲁Licensein Vicineum
上皮细胞黏附分子(EpCAM, EpithelialCellular adhesion molecule)基因在上皮组织中广泛表达,在人胚胎期除了胸腺,几乎所有上皮组织均表达EpCAM基因。出生后,除了鳞状上皮和干细胞外,其他上皮仍维持EpCAM基因的表达。EpCAM蛋白参与加快细胞周期、促进细胞增殖、分化、迁移以及免疫逃逸等多种生物学功能,在肿瘤学中,“干细胞”学说认为肿瘤干细胞是肿瘤中具有自我更新能力并能产生异质性肿瘤细胞群的细胞,其与肿瘤的增殖、转移、复发和对放疗不敏感关系密切。EpCAM作为肿瘤干细胞标志物,在消化系统肿瘤中较为常见,尤其是与肝癌、结直肠癌、胃癌等关系十分密切。
尽管EpCAM是目前表达最强和最广泛的肿瘤表面抗原之一,前景令人瞩目,但是EpCAM涉及多基因变化的一系列复杂过程,其表现形式和调节方式也复杂多样,尚有待深入研究。此外,EpCAM抗体具有特异性较差和结合力较低等弊端,临床试验结果不甚理想,目前研究还处于初级阶段。
(1)Opotuzumab manatox
Sesen-Bio是一家位于美国马萨诸塞州的生物制药公司,其研发的Vicineum(oportuzumab monatox)是由重组人源化抗EpCAM抗体oportuzumab与假单胞菌外毒素A偶联而成的ADC药物。在临床前研究证实,EpCAM在非肌层浸润性膀胱癌(NMIBC)细胞中过度表达,但在正常膀胱细胞中几乎无表达。
Linker:Vicineum由一个稳定的基因工程肽链构成,确保免疫毒素保持附着状态直至被癌细胞内化,从而降低对健康组织的毒性风险。
Payload:Vicineum为一款使用免疫毒素ADC药物。免疫毒素是用于选择性破坏具有某一特异性标记细胞的一类融合蛋白,通常是由具有高度特异性的单克隆抗体与具有强大杀伤作用的毒素分子通过化学交联构建而形成。免疫毒素具有高效、低毒、高特异性等特点,但免疫毒素也存在不少问题,如对靶组织的特异性杀伤作用弱,诱发不同程度的血管渗透综合征(VLS)等。
作用机制:Vicineum具有双重作用机制,一方面,Vicineum内化入EpCAM高表达的肿瘤细胞后,释放免疫毒素杀死靶细胞;另一方面,Vicineum可激活免疫系统,T细胞识别肿瘤新生抗原,杀灭靶细胞。
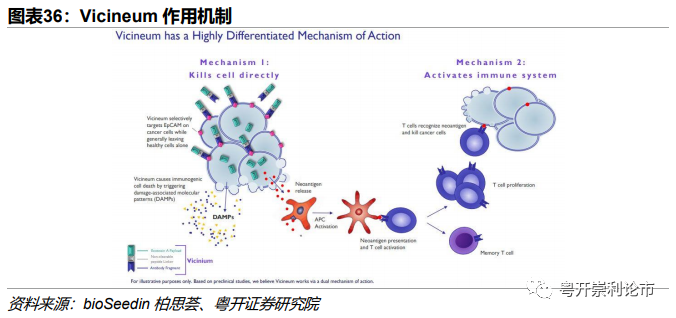
FDA上市申请:2021年2月,Sesen Bio宣布FDA已经受理其创新ADC药物Vicineum治疗对卡介苗(BCG)无应答的高风险NMIBC的上市申请,并授予优先审评资格,PDUFA预定审批期限是2021年8月18日。

非肌层浸润性膀胱癌(NMIBC)
试验设计:Vicineum非肌层浸润性膀胱癌的上市申请基于一项为期24个月的多中心、单臂、开放标签VISTA研究。试验入组133例高级别NMIBC原位癌(CIS)或乳头状癌患者,这些患者曾接受过卡介苗(BCG)治疗,包括3个队列。队列1:BCG治疗6个月内难治或复发CIS;队列2:BCG治疗6-11个月内复发CIS;队列3:BCG治疗6个月内难治或复发乳头状癌。研究中,患者接受局部给药Vicinuem每周2次治疗6周,随后每周一次治疗6周,之后每隔一周治疗一次,持续2年。试验的主要终点是原位癌或不伴乳头状病变患者的完全缓解率和缓解时间。
试验结果
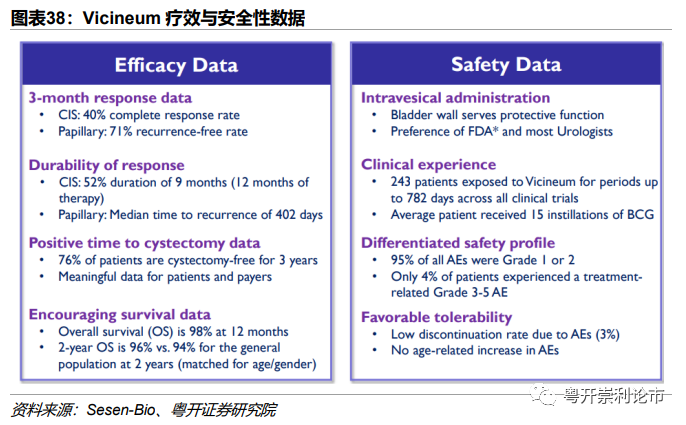
疗效结果:截至2019年5月29日,主要和次要终点数据如下:(1)CR:队列1在第3、6、9、12个月的完全缓解率(CR)为39%、26%、20%和17%,队列2的CR率为57%、57%、43%和14%。(2)mDOR:队列1的mDOR为273天,队列1和队列2所有CIS患者在3个月时间点实现完全缓解的患者中,有52%患者在启动治疗后的完全缓解持续时间≥12个月。(3)疾病复发时间:高危乳头瘤NMIBC疾病复发时间为关键次要终点,队列3的中位疾病复发时间为402天;(4)肿瘤切除术时间:BCG无应答NMIBC治疗目标是避免膀胱切除,结果显示>75%患者2.5年保持膀胱无切除,88%应答者在3年保持膀胱五切除;(5)PFS:90%患者的PFS≥2年;(6)OS:96%患者OS≥2年。
安全性结果:Vicineum表现出良好的耐受性,95%的TRAE为1级或2级,最常见的治疗相关不良事件为排尿困难(14%)、血尿(13%)和尿路感染(12%)
结论:Vicineum对非肌层浸润性膀胱癌疗效显著,可有效促进抗肿瘤免疫反应。除了单药治疗NMIBC外,Vicineum正在进行与阿斯利康的度伐利尤单抗联用的临床试验,探索治疗高危、BCG无反应NMIBC的疗效,目前处于Ⅰ期临床(NCT03258593)。
(2)齐鲁制药
2020年7月31日,齐鲁制药与Sesen Bio公司达成独家授权协议,获得Vicineum在大中华区的独家开发和商业化权益,首付款为1200万美元,后续里程碑费用合计2300万美元。Vicineum在大中华商业化后,Sesen Bio有权获得基于大中华净销售额的特许权使用费。2021年3月19日,Vicineum(莫奥珠单抗,VB4-845)已默认获批临床,拟用于非肌层浸润性膀胱癌。
(二)Nectin-4ADC:Padcev尿路上皮癌后线新疗法
Nectin-4是一种Ⅰ型膜蛋白,在正常的胚胎和胎儿组织中含量很高,成年后下降,在建康组织中的分布有限。Nectin-4在多种肿瘤细胞中过度表达,如尿路上皮癌、乳腺癌、胰腺癌、三阴乳腺癌和膀胱癌等,可通过激活PI3K/Akt途径促进肿瘤细胞增殖、分化、迁移、侵袭等。尽管靶向Nectin-4的ADC药物Padcev已于2019年在美国加速获批上市,但与HER2、CD22等热门靶点相比,目前针对Nectin-4靶点布局的ADC产品较少,Nectin-4靶点未来开发潜力较大。
Enfortumab Vedotin商品名为Padcev,是由Seattle Genetics和安斯泰来联合开发。Padcev是由靶向Nectin-4的人IgG1单抗Enfortumab与微管蛋白抑制剂MMAE偶联而成的抗体偶联药物(ADC)。Padcev适应症为尿路上皮癌。尿路上皮癌占所有膀胱癌的90%,在接受含铂初始化疗失败后,80%的晚期患者对PD-1/L1抑制剂治疗难以产生应答,目前,这部分患者有了新的治疗药物。
FDA批准:2019年,Padcev获FDA批准上市,用于治疗之前接受过PD-1/PD-L1抑制剂或含铂类化疗的成人局部晚期或转移性尿路上皮癌患者。
NMPA递交临床申请:2020年12月14日,安斯泰来在中国提交的Padcev临床试验申请获得审批,用于治疗局部晚期或转移性尿路上皮癌。
作用机制:Padcev与表达Nectin-4的癌细胞结合后,内化入靶细胞,在溶酶体中降解,释放细胞毒素MMAE,MMAE与微管蛋白结合,导致细胞周期停滞,使细胞凋亡。

(1)尿路上皮癌(UC)
队列1实验设计:Padcev获得FDA的加速获批上市基于单臂、双队列、关键性Ⅱ期临床研究EV-201队列1的结果。队列1入组之前曾接受PD-1/PD-L1抑制剂治疗和含铂化疗的患者,队列2入组之前接受过PD-1/L1抑制剂治疗、未接受铂类化疗且不符合顺铂治疗条件的患者。队列1试验纳入125名既往接受过PD-1/PD-L1抑制剂和含铂化疗方案治疗的局部晚期或转移性尿路上皮癌患者,所有患者在每个28天周期的第1天、第8天和第15天静脉注射1.25mg/kg Padcev。试验的主要终点为客观缓解率(ORR),次要终点包括反应持续时间(DOR)、无进展生存期(PFS)、总体生存期(OS)和安全性/耐受性。
实验结果
疗效数据:试验结果显示,Padcev治疗迅速缩小了大多数患者的肿瘤,客观缓解率(ORR)为44%(55/125,95%CI:35.1、53.2),完全(CR)和部分缓解率(PR)分别为12%和32%,28%的患者SD,中位PFS为5.8个月,中位OS为11.7个月,中位DOR为7.6个月。
安全性数据:常见治疗相关不良事件(TRAEs)包括疲劳(50%)、脱发(49%)和食欲下降(44%)。12%的患者因TRAE而停止治疗,最常见的原因是周围神经病变。

队列2实验设计:队列2试验为一项开放标签、多中心、全球试验,入组89名未接受铂类化疗且不符合顺铂治疗条件的患者。所有患者在每个28天周期的第1、8和15天接受1.25mg/kg的Padcev治疗。试验的主要终点为客观缓解率(ORR),次要终点为DOR、PFS、OS与安全性数据。
实验结果
疗效数据:试验结果显示,客观缓解率(ORR)为52%(95%CI:40.8、62.4),完全缓解率(CR)达20%,中位DOR达10.9个月,中位PFS为5.8个月,中位OS为14.7个月。我们认为,针对之前接受过PD-1/L1抑制剂治疗、未接受铂类化疗且不符合顺铂治疗条件的晚期或转移性尿路上皮癌患者,Padcev高达20%的CR率和良好的PFS、OS结果令人鼓舞。
安全性数据:常见治疗相关不良事件(TRAEs)包括脱发(51%)、周围神经病变(47%)和疲劳(34%)。特别关注的治疗相关AE包括皮疹(所有等级61%,≥3级17%)、周围神经病变(所有等级54%,≥3级8%)和高血糖(所有等级10%,≥3级6%)。研究者报告了4例与治疗相关的死亡,均发生在≥75岁且有多种合并症的患者中:3例死亡事件(急性肾损伤、代谢性酸中毒和多器官功能障碍综合征)发生在首次EV给药的30天内BMI≥30的患者中,1例死亡事件(肺炎)发生在最后一次给药的30天后。
(2)与Keytruda联合治疗尿路上皮癌:获批突破性药物资格
2020年2月,FDA批准Padcev与Keytruda联合用药方案突破性药物资格(BTD),用于治疗在一线治疗种无法接受顺铂化疗的局部晚期或转移性尿路上皮癌(UC)患者。此次突破性药物资格基于EV-103试验剂量递增队列和扩大队列A的结果,Padcev与Keytrude联合用药方案在尿路上皮癌患者治疗时显示出良好的效果。
实验设计:EV-103是一项多队列、开放标签、多中心Ⅰb/Ⅱ期临床试验,旨在评估Padcev+Keytruda方案治疗肌肉浸润性、局部晚期、一线和二线转移性尿路上皮癌的安全性、耐受性和疗效。
实验结果
疗效数据:结果显示,Padcev+Keytrude确认的客观缓解率(ORR)为73.3%(n=33/45,95%CI:58,85.4);其中,完全缓解率(CR)为15.6%(n=7/45),部分缓解率(PR)为57.8%(n=26/45);中位缓解持续时间(DOR)尚未达到;中位无进展生存期(PFS)为12.3个月;12个月总生存率(OS)为81.6%,中位OS尚未达到。
(三)Claudin18.2:康诺亚独家布局
Claudin是人体正常组织中紧密连接最重要的一种蛋白质,具有4个跨膜结构域,是细胞紧密连接的重要分子,参与集体生理过程如细胞旁通透性和电导的调节,其构成了细胞旁屏障。该家族拥有至少24个成员,不同的Claudin蛋白表达于不同的组织,与各自组织肿瘤发生具有相关性,其中Claudin-1与结直肠癌高度相关,Claudin-10与肝细胞癌发生密切,Claudin-18在胃癌上高表达。
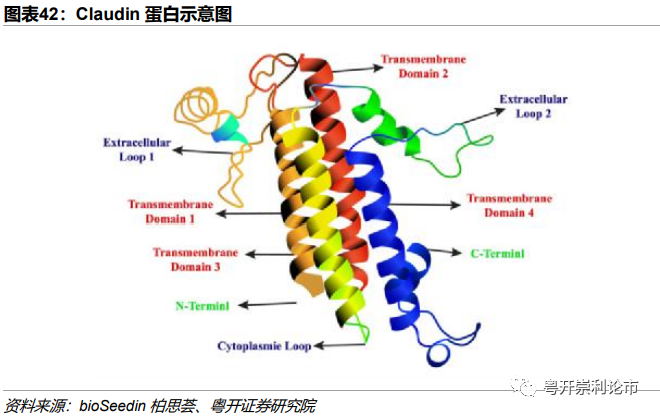
Claudin-18包含Claudin-18.1和Claudin-18.2两种异构体。在正常组织中,Claudin-18.2仅在分化的胃粘膜上表皮细胞中表达,但是在原发性胃癌、转移性胃癌、胰腺癌、食管癌、卵巢癌、肺癌等多种实体瘤中常常观察到Claudin-18.2被激活表达,例如50%-80%的胃癌患者中存在Claudin-18.2的表达。这种在正常细胞无/低表达、在癌细胞高表达的特性使Claudin 18.2近年来成为实体瘤免疫疗法的理想靶点,受到研究人员的高度关注。
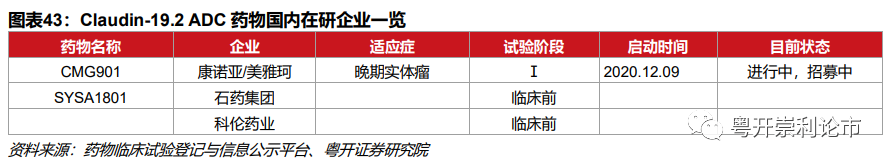
目前康诺亚/美雅珂布局Claudin-18.2的ADC药物CMG901,目前处于计量探索和计量扩展Ⅰ期临床研究阶段,适应症为无标准治疗的晚期实体瘤。石药集团的Claudin 18.2ADC药物SYSA1801于2020年11月获得FDA的孤儿药资格认定,计划于2021年递交中国、美国临床试验申请。科伦药业的Claudin 18.2 ADC药物预计2021年进入临床。
六、国内主要ADC药物研发企业
受全球创新浪潮的冲击和ADC药物的更新迭代,国内ADC药物的研发热情高涨,一批生物医药企业相继奔赴ADC药物创新的战场。除了荣昌生物、浙江医药等重点布局HER2 ADC产品,云顶新耀、科伦药业、君实生物布局了Trop2 ADC产品,康诺亚/美雅珂布局了Claudin 18.2 ADC产品,恒瑞医药、荣昌生物深耕于cMET ADC产品。
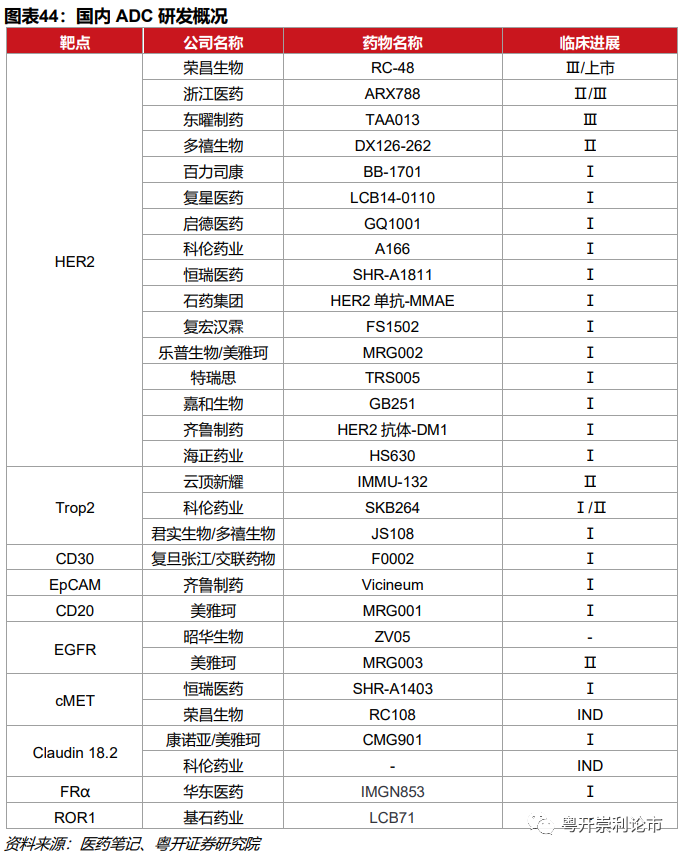
云顶新耀:2019年4月,云顶新耀获得Trodelvy在大中华区、韩国及部分东南亚国家的独家权益,涉及金额高达8.35亿美元。2020年5月,Trodelvy获NMPA批准开展针对mTNBC的Ⅰ期临床。2020年12月9日,云顶新耀公告Trodelvy已完成用于转移性乳腺癌(mBC)亚洲3期临床试验首例患者给药。
科伦药业:SKB264是科伦博泰自主研发的TROP-2 ADC药物,含有三个主要组成部分:1)重组抗Trop2人源化单克隆抗体;2)含2-(甲基磺酰基)嘧啶接头的连接子;3)拓扑异构酶抑制剂KL610023。2020年4月9日,科伦博泰的SKB264获得国家药监局临床试验通知书。目前,SKB264处于Ⅰ/Ⅱ期临床。
君实生物/多禧生物:JS108为注射用重组人源化抗Trop2单抗-Tub196偶联剂,用于治疗三阴乳腺癌、肺癌和胰腺癌。2019年12月,君实生物通过独占许可授权方式从多禧生物获得JS108的许可使用权,双方约定3000万元的首付款、不超过2.7亿的研发和上市里程碑费用,以及年销售收入6%-10%的收益分成。2020年7月21日,JS108获NMPA核准签发的《药物临床试验批准通知书》,用于晚期实体恶性肿瘤的临床试验。2020年11月25日,上海君实生物的JS108的1期临床研究已完成首例患者给药。
恒瑞医药:恒瑞医药布局了多款ADC药物,其中靶向HER2的ADC药物SHR-A1811处于Ⅰ期临床,靶向cMet的ADC药物SHR-A1403目前处于临床Ⅰ期。
康诺亚/美雅珂:康诺亚/美雅珂布局Claudin-18.2的ADC药物CMG901,目前处于计量探索和计量扩展Ⅰ期临床研究阶段,适应症为无标准治疗的晚期实体瘤。
齐鲁药业:齐鲁制药与Sesen Bio公司达成独家授权协议,获得Vicineum(EpCAM ADC)在大中华区的独家开发和商业化权益,首付款为1200万美元,后续里程碑费用合计2300万美元。Vicineum在大中华商业化后,Sesen Bio有权获得基于大中华净销售额的特许权使用费。
荣昌生物-B:荣昌生物深耕于ADC领域,靶向HER2的ADC药物RC48已于2020年8月向NMPA递交晚期胃癌适应症的上市申请。RC88为针对间皮素的ADC药物,目前处于临床Ⅰ期;RC108为靶向cMet的ADC药物,目前处于IND阶段。
华东医药:2020年10月20日,华东医药与美国ImmunoGen,Inc.达成独家临床开发及商业化协议。ImmunoGen授予华东医药其卵巢癌三期临床在研ADC产品Mirvetuximab Soravtansine在中国大陆、香港、澳门和台湾地区临床开发和商业化权益。交易总金额超过3亿美金,其中包括4000万美元的首付款和2.65亿美元的潜在里程碑付款。
七、风险提示
药物研发风险、药品降价风险、上市后销售不及预期。
粤开证券研究院策略&行业团队
粤开证券研究院策略及行业团队具备完善的研究体系,观点鲜明,是高效且紧贴市场的实战型团队。2020年,策略与行业团队凭借专业及深度研究,赢得了市场的认可和尊敬。2021年,粤开证券研究院在大股东广州开发区控股的引领下将整装再出发,立足粤港澳大湾区,辐射全中国,造就一流特色精品研究!


免责声明:自媒体综合提供的内容均源自自媒体,版权归原作者所有,转载请联系原作者并获许可。文章观点仅代表作者本人,不代表新浪立场。若内容涉及投资建议,仅供参考勿作为投资依据。投资有风险,入市需谨慎。






APP专享直播
热门推荐
收起
24小时滚动播报最新的财经资讯和视频,更多粉丝福利扫描二维码关注(sinafinance)







