
新浪财经ESG评级中心提供包括资讯、报告、培训、咨询等在内的14项ESG服务,助力上市公司传播ESG理念,提升ESG可持续发展表现。点击查看【ESG评级中心服务手册】
基于生物质基碳纳米片的水污染物氧化脱除
郝熔江1,2,3,顾翔宇1,李松庚1,2,4*
1.中国科学院过程工程研究所,介科学与工程全国重点实验室,北京 100190
2.中国科学院大学中丹学院,北京 100049
3.中国丹麦科研教育中心,北京 100049
4.中国科学院大学化学工程学院,北京 100049
摘要:通过在熔融盐辅助热解法中加入含氧酸盐(K2CO3或 KNO3),并以葡萄糖为碳源成功合成了高石墨化和多缺陷位点的多孔二维碳纳米片 M-C和 M-N.含氧酸盐显著提高了纳米片的比表面积,特别是 KNO3还促进氮掺杂,使 M-N 对酸性橙 7(AO7)的最大吸附容量达到 480.77mg/g,远高于直接热解生物炭 BC 和无含氧酸盐的熔融盐热解炭 M-BC.碳材料对 AO7 去除的吸附和催化降解过程具有协同效应,M-N 活化过一硫酸盐(PMS)的活性分别为 M-BC 的 22.64 倍和 BC 的 33.48 倍.此外,通过密度泛函理论(DFT)初步计算了氮掺杂和不同缺陷对非自由基主导的催化过程的影响.本研究发现含氧酸盐可以明显降低碳纳米片制备过程中熔融盐添加量,也为开发有机污染物高效吸附和 PMS 高效活化的生物质基双功能碳材料提供了理论指导.
关键词:碳纳米片;热解;吸附;催化剂;高级氧化
染料废水作为典型的工业废水,已引起严重的环境问题,特别是对水生环境的污染.以-N=N-键为特征的不可生物降解偶氮染料占总染料产量 50%以上[1],这类染料在环境中的积累会对生物体产生严重的致癌、致畸和致突变风险.因此,开发高效且低成本的水处理技术受到广泛关注[2].目前已报道多种从水中去除偶氮染料的方法,如吸附[3]、微生物降解[4]、光催化降解[5]和化学氧化[6]等,其中,吸附法操作最为简单,但仅能富集污染物,不能对其降解,需要进行后处理以减少二次污染.
目前,基于过一硫酸盐(PMS)的高级氧化过程(AOP)已成为一种有前景的降解废水中难降解有机污染物的方法[7-9].PMS 的活化是处理过程中的关键步骤,可通过加热、紫外线照射、超声波和催化剂等实现[10-11].其中,非金属碳基催化剂因其独特的电子性质、可调的结构特性、良好的生物相容性以及相对低廉的成本成为研究热点.相较于热、紫外线和超声波活化,非金属碳基催化剂可以避免大量能量输入,并减少因使用过渡金属催化剂引起的有毒金属浸出问题,降低二次污染风险[2,12-13].
有研究表明,有机污染物在碳材料上的吸附可缩短其与活性物质间的距离,从而促进降解[14].以石墨烯及其衍生物为代表的二维碳材料,凭借其高比表面积和独特的 sp2杂化碳构型,通过 π−π 相互作用对具有苯环结构的有机污染物表现出强吸附性[15].此外,二维碳材料充分暴露的活性位点、高电荷载流子迁移率和高导电性有利于提升其催化活性[16].研究发现,在活化过硫酸盐降解亚甲基蓝和苯酚的过程中,二维还原氧化石墨烯(rGO)相比其它维度的碳材料表现出更高的效率[17].因此,开发兼具吸附和催化双功能的二维碳材料是降解水中有机污染物的关键.然而,目前用于过硫酸盐高级氧化的二维碳材料主要依赖于高成本的石墨烯作为碳前驱体进行改性以提高其催化性能[14, 16],这限制了实际大规模应用的可行性.对常见的含碳有机物进行熔融盐辅助热解是合成二维石墨烯基碳材料的有效方法,但研究发现,为实现高比例的二维结构和高孔隙率,需使用高达 1:100 的葡萄糖与氯化钾/氯化锂熔融盐质量比[18],增加了材料合成前处理和后处理的复杂性,提高了合成成本.有研究发现,在sp2杂化碳的热解制备过程中,其倾向于堆叠形成热力学稳定的致密石墨结构,而引入氧化性的含氧酸盐可促进碳源分解,使其转化为高孔隙率的碳纳米结构[19].
基于此,本研究通过在葡萄糖熔融盐辅助热解体系中分别引入两种含氧酸盐,包括KNO3和K2CO3,探索在较低熔融盐用量下,高性能二维碳基催化剂的制备方案.以酸性橙 7(AO7)为目标污染物,深入探究了所制备碳材料的吸附和催化特性,揭示了二维结构和氮掺杂对性能的影响,从而为生物质基二维碳催化剂的简便合成及其在有机污染物吸附和PMS 活化过程的应用提供指导.
1 材料和方法
1.1 主要实验试剂
无水葡萄糖、氯化钾(KCl)、氯化锂(LiCl)、碳酸钾(K2CO3)、硝酸钾(KNO3)、过硫酸氢钾复合盐(2KHSO5·KHSO4·K2SO4),亚硝酸钠(NaNO2)、甲醇、叔丁醇、二甲基亚砜、对苯醌、2,2,6,6-四甲基哌啶(TEMP)、5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)购买自上海麦克林生化科技股份有限公司;酸性橙7、糠醇购买自阿拉丁(14.720, -0.23, -1.54%)试剂有限公司.上述试剂均为分析纯.实验用水为去离子水.
1.2 碳材料的制备
采用热解法制备碳材料.将无水葡萄糖、含氧酸盐 (K2CO3 或 KNO3) 和 共 晶 熔 融 盐 KCl/LiCl(KCl:41mol.%)按照 1:1:10 的质量比充分混合.然后将混合物放入刚玉瓷舟中,并在管式炉(QSK 1200)中进行热解,处理时间为 3h,热解温度设定为 800 , ℃气氛为氮气.完成热解后,待样品自然冷却至室温,再将热解产物研磨成粉末,并将其分散在去离子水中.通过过滤并在 60℃下真空干燥 24h,以得到最终的热解碳材料.根据使用的含氧酸盐不同,所得碳材料分别命名为 M-C(使用 K2CO3)和 M-N(使用 KNO3),其中“M”表示熔融盐辅助热解;在熔融盐环境中不添加任何含氧酸盐情况下得到的热解炭记为 M-BC;使用葡萄糖直接热解得到的热解炭记为 BC.
1.3 表征仪器与方法
使用扫描电子显微镜(SEM, JSM-7800F(Prime))和透射电子显微镜(TEM, JEM-F200)分析碳材料的形貌结构,使用和 SEM 配套的能谱仪(EDS)分析样品的表面元素组成及分布.采用 X 射线源为 Cu Kα的 X 射线衍射仪(XRD, SmartLab 9kW)对材料的物相和晶体结构进行表征.利用 BET 比表面全自动物理吸附仪(Autosorb IQ)测定样品的孔结构.通过X射线光电子能谱仪(XPS, ESCALAB 250Xi)分析样品表面元素组成和含量,以 C 1s 峰位为 284.8eV 进行荷电校正.通过激光拉曼光谱仪(Raman, LabRAMHP Evolution)分析碳材料的石墨化程度和结构缺陷程度等 信 息 . 使用电子 顺磁共振 波 谱 仪 (EPR,EMXplus-9.5/12)鉴定催化反应体系内产生的活性氧化组分(ROSs).
1.4 碳材料吸附和催化性能测试
首先对碳材料的吸附性能进行探究.实验流程如下:在锥形瓶中配制200mL初始浓度为20mg/L的AO7 溶液,将 30mg/L 的 BC、M-BC、M-C 或 M-N样品投加到溶液中,置于振荡频率为 240r/min 的气浴恒温摇床中开始吸附实验,溶液初始 pH 值为 5.0,实验温度为(25.0±0.5) . ℃ 到达取样时间后,吸取 1mL溶液并使用 0.45µm 的水系针头过滤器除去残留的碳材料,收集滤液测定其浓度.
在研究碳材料的催化性能时,先进行吸附实验等待吸附-解吸平衡后,再将 1.0mmol/L 的氧化剂PMS 添加到溶液中以引发催化降解反应.到达预定的时间间隔后,取样 1mL 溶液,并快速与 2mL 的NaNO2 溶液(0.1mol/L)混合以淬灭反应,接着使用0.45µm 的水系过滤器除去碳催化剂,收集滤液对其浓度进行测定.此外,在新配制的 AO7 溶液中只添加PMS 进行降解实验,以确定 PMS 自活化对 AO7 去除的贡献.吸附和催化实验均重复 3 次,结果取平均值,以减少实验误差.
AO7 的浓度使用紫外-可见光分光光度计(UV-Vis, UV5Bio)在 484nm 处进行测定,其去除率依据式(1)计算;碳材料对 AO7 的吸附容量按式(2)计算.

1.5 EPR 测试
配制 100mmol/L 的 DMPO 水溶液,从催化剂/PMS 体 系 (AO7:20mg/L, 催化剂 :30mg/L,PMS:1.0mmol/L,体积:200mL)中提取 1mL 样品,使用0.45µm的水系过滤器除去碳催化剂,将40µL的过滤液与40µL的DMPO溶液混合,通过EPR检测DMPO混合液中加合物 DMPO-⋅OH 和 DMPO-SO4·− 的信号.类似地,使用 100mmol/L 的 DMPO 甲醇溶液捕获O2·− ,使用 100mmol/L 的 TEMP 水溶液捕获 1O2,然后通过 EPR 测试分别检测 DMPO- O2·− 和 TEMP-1O2信号.
2 结果与讨论
2.1 碳材料的表征
2.1.1 形貌和物相结构表征
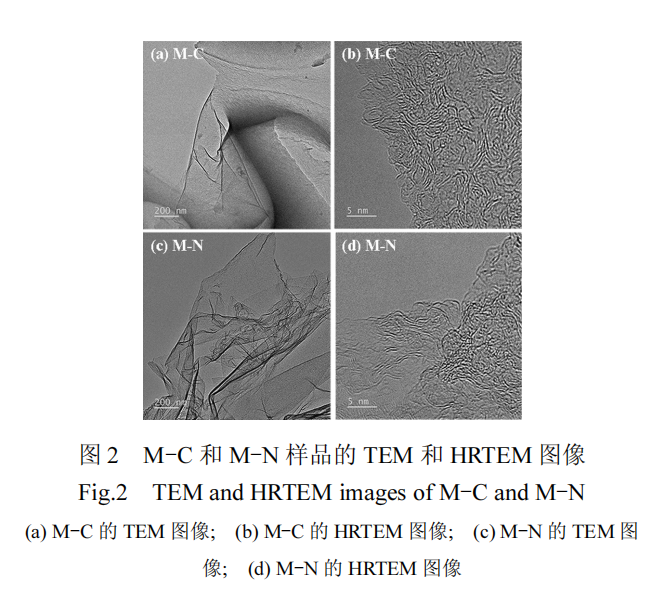
采用 SEM 分析对各样品形貌进行表征,结果如图 1(a)~图 1(f)所示.直接热解的 BC 样品显示出大块碳颗粒的堆积形态.引入熔融盐后,M-BC 样品展现出不规则的碳颗粒与少量层状结构的混合形态.这表明熔融盐体系在热解过程中对热解前驱体的“溶解-沉积”作用可有效抑制葡萄糖直接热解时的碳颗粒聚集现象,促进二维片层结构的生成.该作用依据热解温度的不同可分为两个阶段:温度低于熔融盐熔点(353 ) ℃ 时,与热解前驱体均匀混合的固态熔融盐起到热解硬模板的作用,调控生物炭的形貌,使其形成被熔融盐阻隔、包覆的带有大孔的碳材料;温度高于熔融盐熔点后,高能离子熔体刻蚀并剥离原有结构,使硬模板作用下生成的薄壁碳转变为层状结构[20-21].因此,生成以二维结构为主的碳材料需要高比例的熔融盐辅助热解过程,这不利于过程的经济性[18].而在熔融盐热解体系内引入含氧酸盐后,碳材料的形貌发生了显著变化.具体而言,在碳酸盐(K2CO3)和熔融盐的作用下,M-C 样品主要形成了大量的微米级片层结构,而不规则碳颗粒的数量显著减少;在硝酸盐(KNO3)和熔融盐体系中,M-N 样品则展现了较少的碳颗粒和更发达的片层结构,这些片层的径向尺寸明显大于 M-C 样品,表明片层结构得到了充分的发展.通过如图 1(g)(h)的 EDS 分析,确认 M-N 样品中成功掺杂了氮元素

此外,M-C 和 M-N 样品的 TEM 和高分辨TEM(HRTEM)图像如图 2 所示,两种样品均呈现出富含褶皱的类石墨烯片层结构,且无序的晶格结构说明片层结构缺陷丰富.因此,含氧酸盐 K2CO3 或KNO3 加入到熔融盐体系中起到了片层结构促进剂的作用,显著促进了较低熔融盐比例条件下高缺陷片层结构的形成.特别地,KNO3 还具有对碳材料进行杂原子氮掺杂的功能,可增强材料的吸附和催化性能[22].
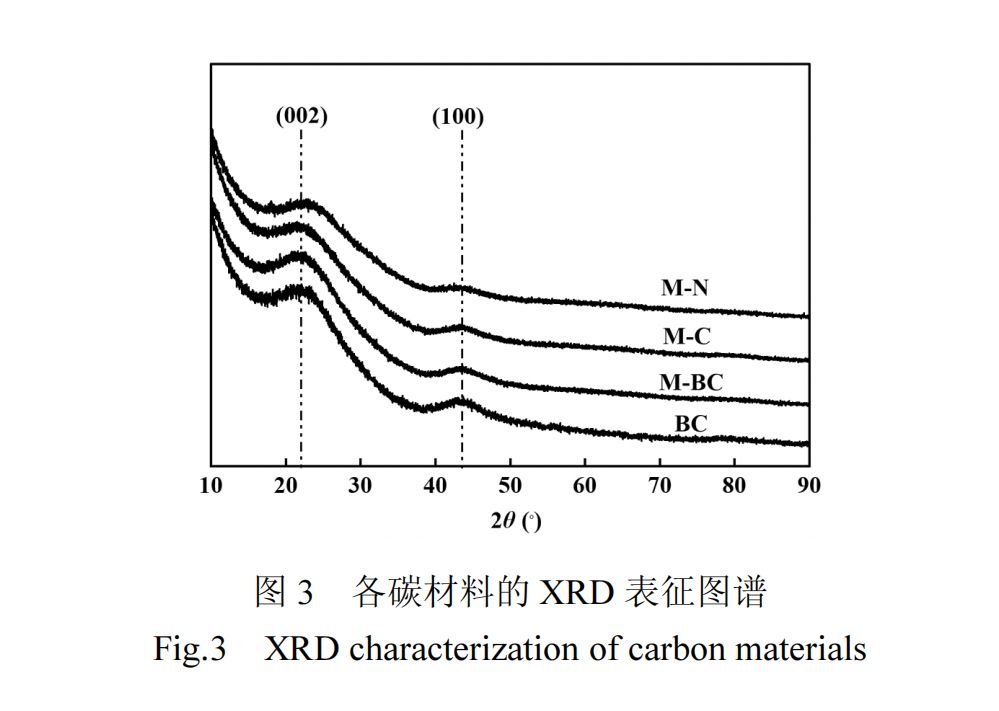
XRD 分析结果如图 3 所示,所有样品均含有两个宽衍射峰,分别对应于石墨的(002)和(100)晶面,这说明碳材料具有堆叠的 sp2 结构.M-BC、M-C 和M-N样品的XRD图谱中无其它衍射峰,进一步证明了经过简单水洗处理,碳材料中的无机盐即可被彻底除去.此外,加入含氧酸盐后,样品的(002)晶面峰向更大的衍射角度移动,表明M-C和M-N样品中石墨微晶的层间距(d002)有所减小[23].这一现象说明含氧酸盐的引入促进碳材料向高石墨化度转变.
2.1.2 孔结构的表征
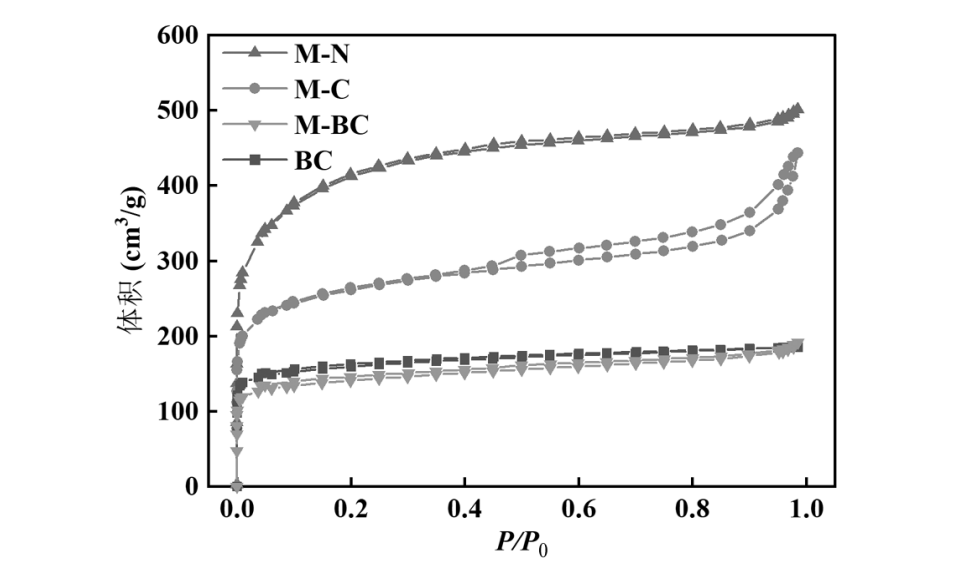
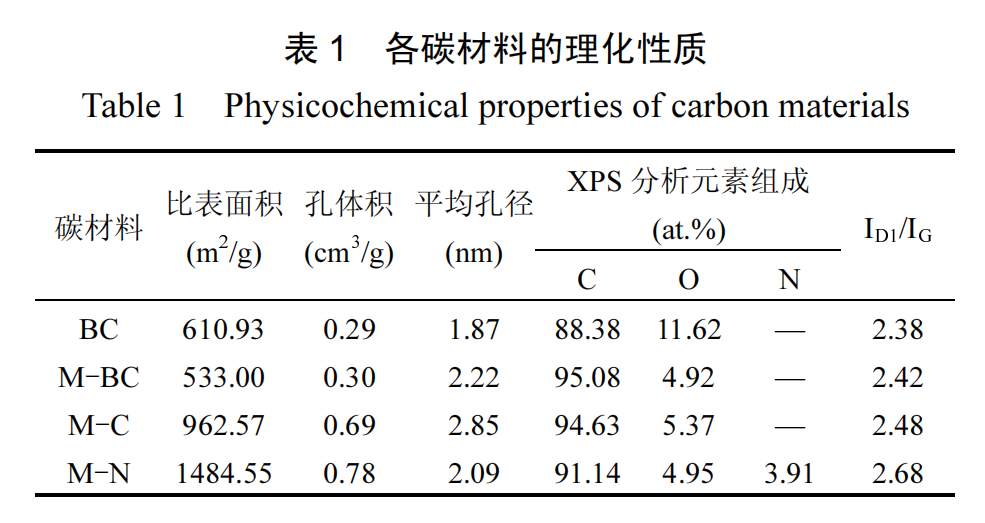
通过氮气吸附-脱附测试评估了碳材料的孔结构.图 4(a)展示了不同样品的吸脱附等温线,它们均呈现 IV 型等温线的特征.通过图4(b)的孔径分布曲线分析可知,各样品均在 2nm 以下有明显的峰,并且累积孔体积显示 BC、M-BC 和M-N 样品的孔体积主要由微孔贡献,而 M-C 样品则显示出较大比例的介孔,这也可以从 M-C 样品在吸脱附等温线中更加明显的 H4 型滞回环及较大的平均孔径(表 1)中得到印证.
M-C 和 M-N 的孔结构形成可归因于含氧酸盐阴离子与葡萄糖热解过程中产生的含碳中间体进行的氧化反应,如式(3)和式(4)所示[19].此外,含氧酸盐的分解通常需要较高温度,但将其与熔融盐混合,可促使其在低熔点的熔融盐介质内发生解离(式(5)和式(6)),从而降低含氧酸盐的分解温度[19,24].K2CO3分解释放的 CO2,KNO3 分解释放的 O2,以及两者生成的 K2O 分别与碳反应(式(7)-(9)),进一步促进了碳材料孔结构的发展[24-25].因此,含氧酸盐与熔融盐的混合体系显著促进了碳材料向高孔隙率活性炭的转变.其中,由于硝酸盐具有更强的氧化能力和更低的分解温度,其孔结构活化作用比碳酸盐更为显著.
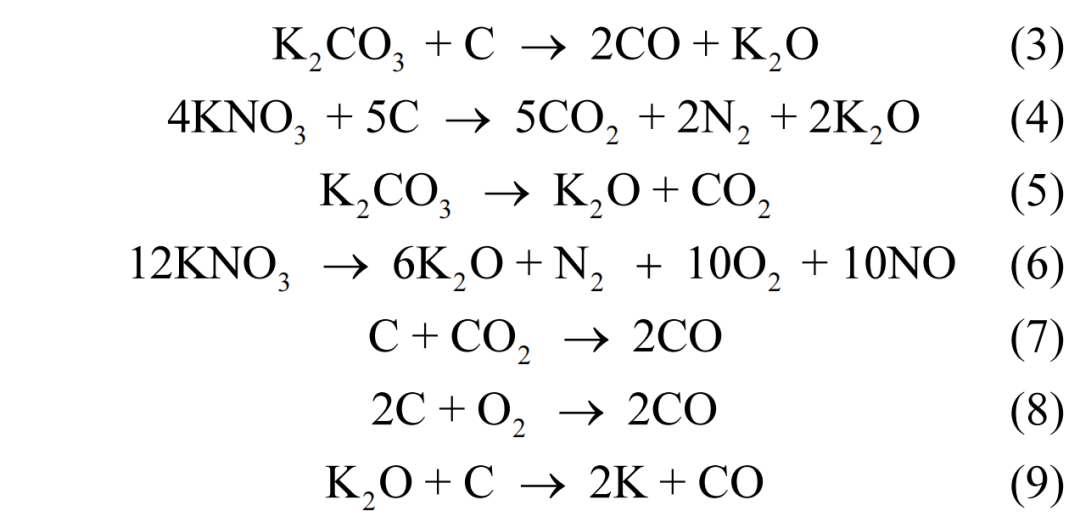
实际上,KNO3 与葡萄糖的混合粉末在 N2 气氛下的热解会引发剧烈的反应,有时甚至导致爆炸[19].而将 KNO3 稀释于熔融盐中,可以显著减缓其对碳材料的氧化反应速率,从而避免爆炸并促进可控的活化反应.这种方法有效促进了孔结构的发展,并确保其在安全可控的条件下进行.
2.1.3 元素组成和分布
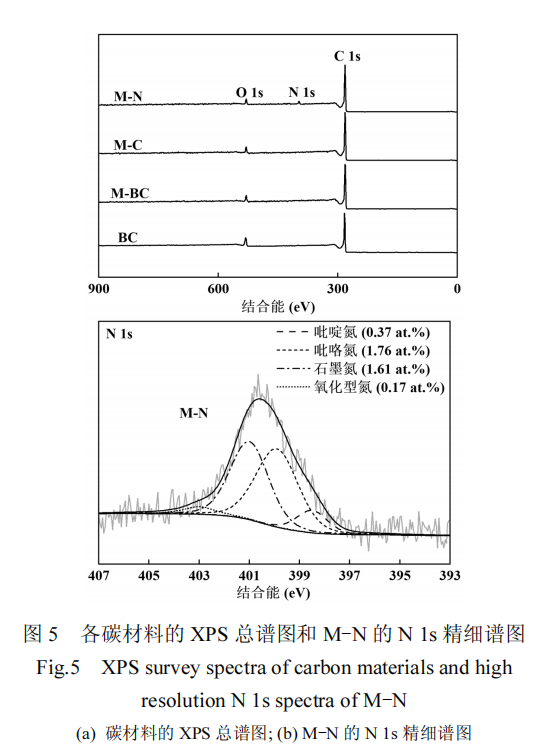
通过 XPS 技术分析了不同样品的表面元素组成及其含量.图 5(a)展示了样品的 XPS 总谱图,从中观察到,所有样品均表现出 C1s 和 O 1s 的特征峰,分别位于 284.8 和 533.7eV,未检测到其它杂质峰,说明样品中的熔融盐已被完全去除.表 1 汇总了不同样品的表面元素组成和含量信息,显示 BC 样品中氧含量最高,引入熔融盐后,熔融盐的刻蚀作用导致 M-BC 中大量的氧被去除.
此外,M-N 样品在 399.8eV 处还出现了 N 1s 的特征峰,其表面元素组成与其它样品相比表现出显著差异.这一结果表明,在 KNO3与碳的反应过程中,氮元素通过碳热还原反应被嵌入到 sp2 杂化的碳结构中,导致 M-N 样品表面出现了氮原子掺杂.因此,在熔融盐体系中引入的 KNO3不仅起到了活化剂的作用,还充当了 N 的来源.
M-N 样品中的 N 1s 高分辨率谱图如图 5(b)所示,区分出几种氮类型:吡啶氮(氮存在于六原子杂环中)、吡咯氮(氮存在于五原子杂环中)、石墨氮(与 3个 sp2杂环碳相邻的 sp2杂化氮)和氧化型氮.这些氮构型结合能分别为 398.5,399.9,401.0 和 403.0eV[20].其中,吡咯氮(1.76at.%)和石墨氮(1.61at.%)为主要存在形式.氮原子掺杂可改善碳材料的电化学特性,从而有效提升对 PMS 的催化活性[26].
2.1.4 结构缺陷表征
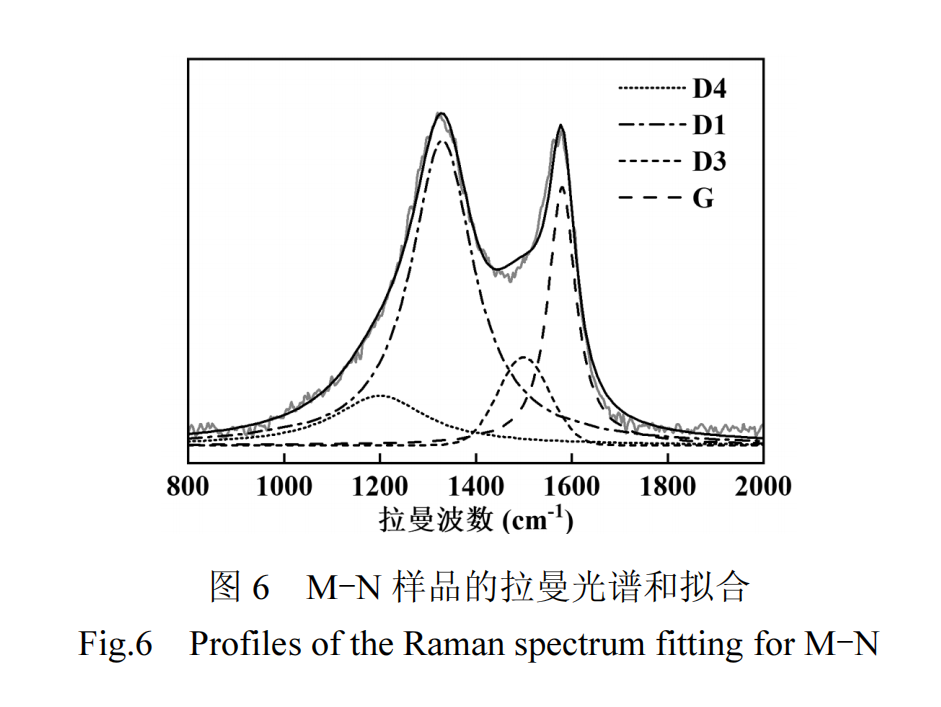
碳材料的缺陷程度会影响其吸附和催化性能,因此使用拉曼光谱仪对样品的结构特性进行研究.以M-N样品为例,其拉曼光谱如图 6 所示,在 800~2000cm-1谱图范围内进行峰拟合,识 别 出 4 个 主要特征 峰 : 洛伦兹 型 的 D4 峰(1200cm-1)、D1 峰(1330cm-1)和 G 峰(1580cm-1)以及高斯型的 D3 峰(1500cm-1)[27-29].通过分析峰值的拟合结果,可得到D1峰和G峰的面积比(ID1/IG),该比值可定量评估碳材料的缺陷程度,反映缺陷石墨晶格中被破坏对称性的碳原子,如锯齿型/扶手椅型边缘缺陷、空位和杂原子掺杂等,其值越大,代表碳材料的无序度和缺陷程度越高[29].
各样品的 ID1/IG值列于表 1,其中 M-BC 的 ID1/IG值相比 BC 显著增加,这归因于熔融盐的引入对缺陷结构生成的促进作用.M-C 和 M-N 样品的 ID1/IG值明显高于 M-BC 样品,表明含氧酸盐和熔融盐混合体系在促进碳材料石墨化转变的同时,在石墨微晶结构中引入了更多的缺陷,包括碳空位、边缘缺陷.此外,M-N 样品中氮原子的引入进一步破坏了石墨晶格中碳原子的对称性,从而显著增加了缺陷程度,使 M-N 样品展现出最高的 ID1/IG值[26].
2.2 碳材料的吸附性能
2.2.1 碳材料的吸附性能评价
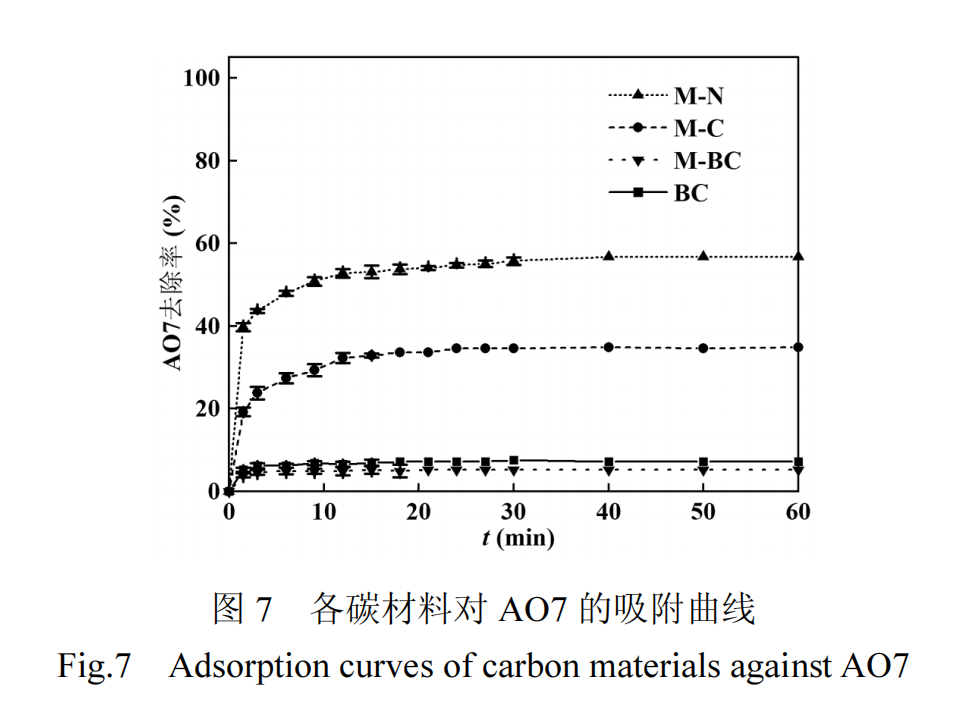
在进行催化性能研究前,首先评估了不同性质的碳材料对 AO7 的吸附性能.如图 7 所示吸附曲线表明,所有碳材料对AO7 的吸附均能在 30min 内迅速达到平衡,且不同材料间的平衡吸附容量存在显著差异,其中 M-N 的吸附能力最强,M-C 次之,均明显优于 M-BC 和 BC.有机物污染物在碳材料表面的吸附主要涉及两种机制:物理吸附和化学吸附.物理吸附主要与碳材料的孔隙结构密切相关,依赖于孔隙的大小和分布.化学吸附则基于有机物与碳材料表面之间的相互作用,这包括:(1)有机物上带负电的芳香环与碳材料缺陷位点之间形成电子供体-受体复合物;(2)有机物芳香环与石墨化碳层之间的 π−π 堆叠相互作用[30].这些相互作用共同决定了有机污染物在碳材料上的吸附能力.因此M-N和M-C样品更强的吸附性能可归因于二者更发达的孔结构、更多的 sp2 碳杂化结构以及更高的缺陷程度,这些特性共同加强了它们对 AO7 的物理和化学吸附.由于 M-N 对 AO7 的吸附性能优于其它样品,因此选择该样品进行进一步的吸附实验研究.
2.2.2 M-N 的吸附动力学
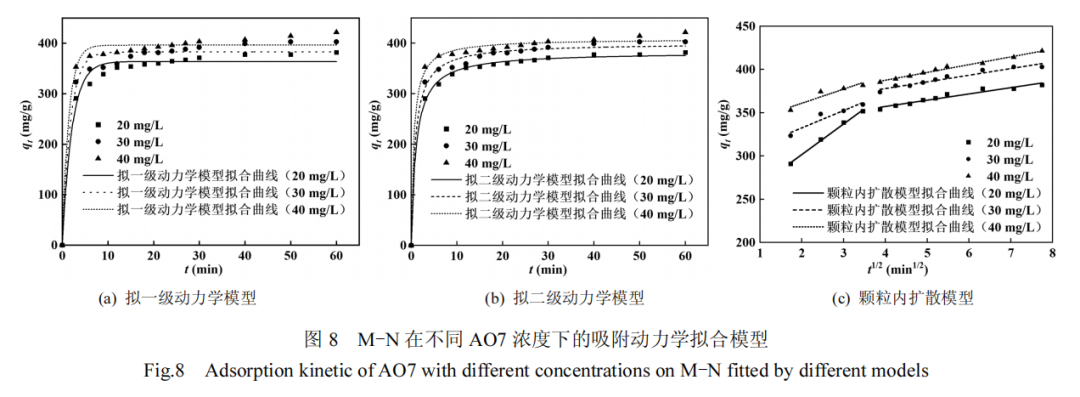
为探究 M-N 样品对AO7 吸附行为的机理,分别使用拟一级动力学模型(式(10))、拟二级动力学模型(式(11))以及颗粒内扩散模型(式(12))对M-N样品吸附不同浓度AO7废水的实验数据进行了拟合.
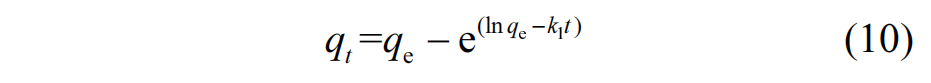

拟合曲线及相应的动力学参数分别见图 8 和表2.分析结果显示,拟二级动力学模型的 R2 高于拟一级模型和颗粒内扩散模型,且其计算得到的饱和吸附容量(qe,cal)与实验值(qe,exp)更为接近,表明该模型更适合描述 M-N 对 AO7 的吸附过程.这说明 AO7在 M-N 上吸附的限速步骤为化学吸附过程,主要受化学相互作用的显著影响[31],包括缺陷位点、吡咯氮和吡啶氮所提供的未成对电子与 AO7 之间的电子供体-受体相互作用,以及石墨晶格与 AO7 分子间的 π−π 堆积作用.颗粒内扩散模型揭示的两个线性阶段指示吸附行为受到双阶段控制.初始阶段为AO7 从溶液主体向 M-N 外表面的扩散,即膜扩散阶段;第二阶段为AO7在M-N孔道结构内部向活性吸附位点的进一步扩散,即颗粒内扩散阶段.通过比较两阶段对应的动力学速率常数 kid,1和 kid,2,进一步表明颗粒内扩散阶段为 M-N 吸附 AO7 过程中的主要限速步骤[32].
2.2.3 M-N 的吸附等温线
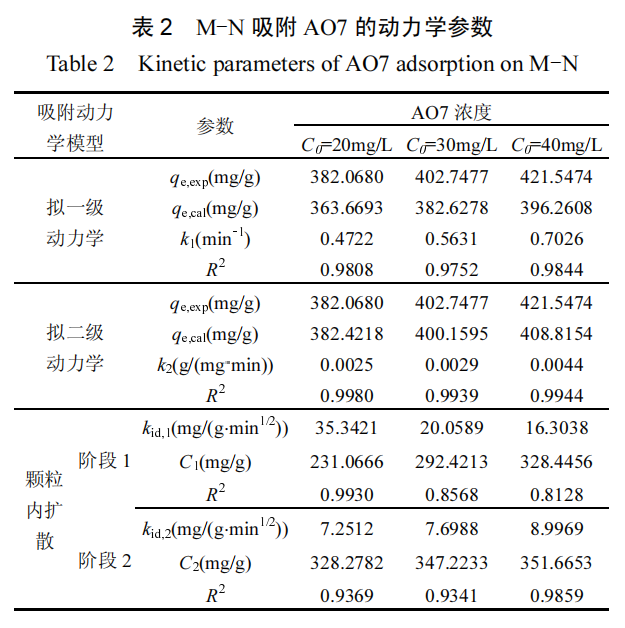
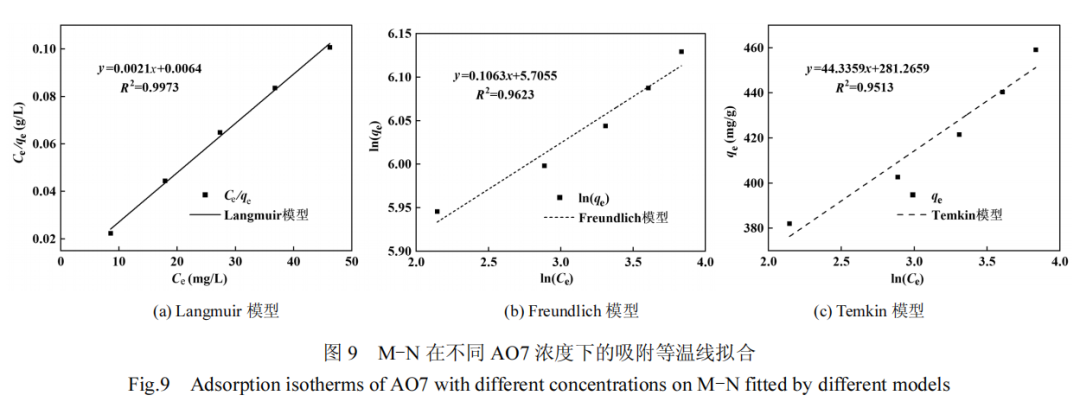
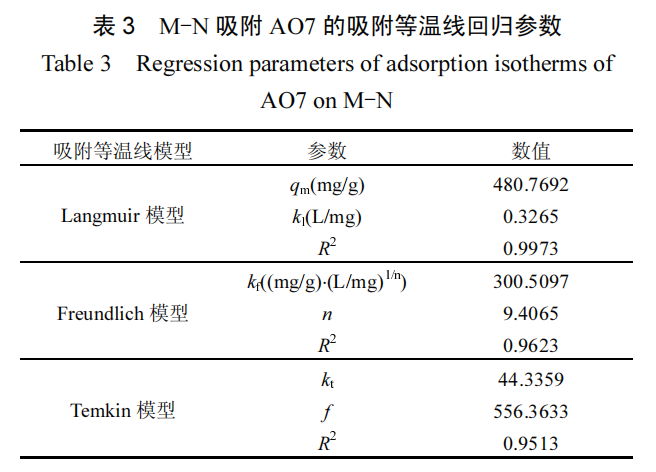
随着 AO7 的初始浓度增加,浓度梯度的作用导致 M-N 的吸附能力呈现逐步增长的趋势.将吸附过程与 3 种经典的吸附等温线 模 型 :Langmuir( 式 (13)) 、 Freundlich( 式 (14)) 和Temkin 等温线模型(式(15))进行拟合.
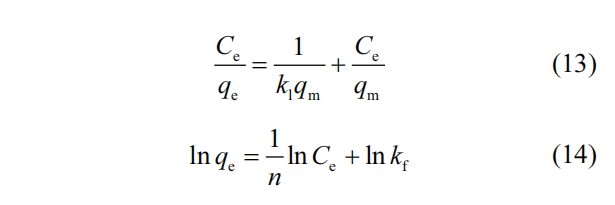

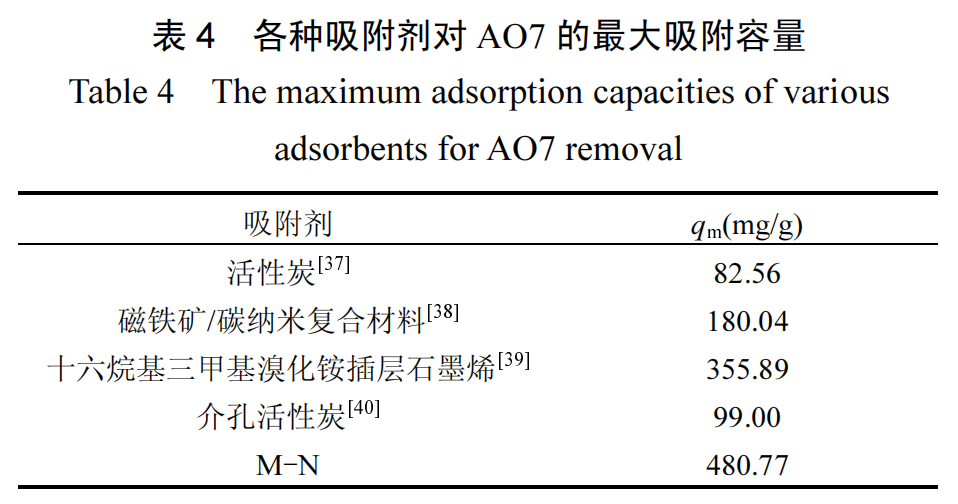
拟合曲线及参数分别见图 9 和表 3.相较于Freundlich模 型 (R2=0.9623) 和 Temkin 模 型(R2=0.9513),Langmuir 模型(R2=0.9973)更能准确描述 M-N 对 AO7 的吸附等温线,指示 AO7 在 M-N 表面均匀分布的吸附位点上发生单层吸附过程[33].Langmuir 模型计算的最大吸附容量为 480.77mg/g,与其它已报道的 AO7 吸附剂相比(表 4),M-N 展现了优异的 AO7 吸附性能.对于 AO7 浓度范围(20~60mg/L),Langmuir 模型计算出分离因子 RL(式(16))的值在 0.0486~0.1328 之间,均落在 0~1 的范围内.这一结果结合 Freundlich 模型中 1/n 小于 1 的结果,均表明 AO7 易于在 M-N 上发生吸附[34-36].
2.3 碳材料的催化性能
鉴于M-N样品对AO7优异的吸附性能,先进行了 30min 的吸附实验以达到吸附平衡,随后引入PMS 启动催化降解过程.此步骤来排除吸附作用对催化过程中 AO7 浓度降低的贡献,确保测得的 AO7去除率准确反映催化降解效果[22].
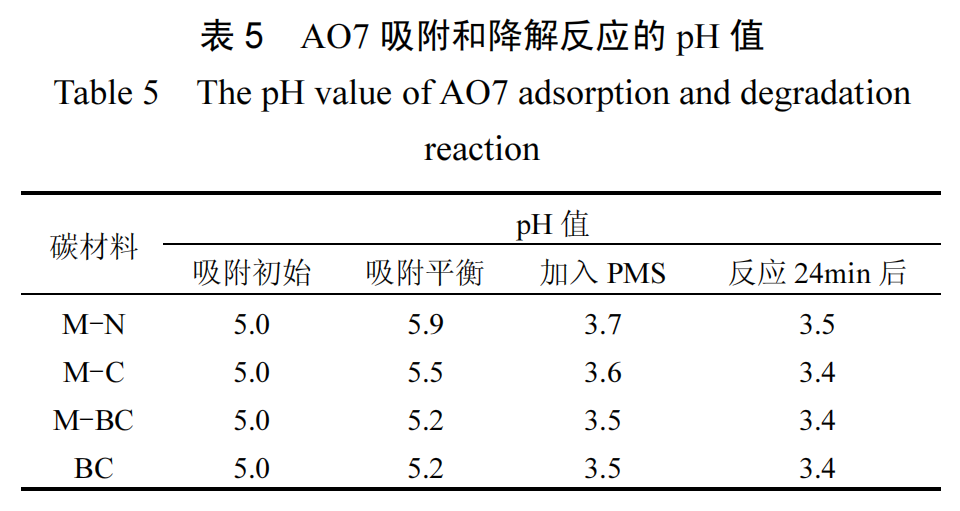
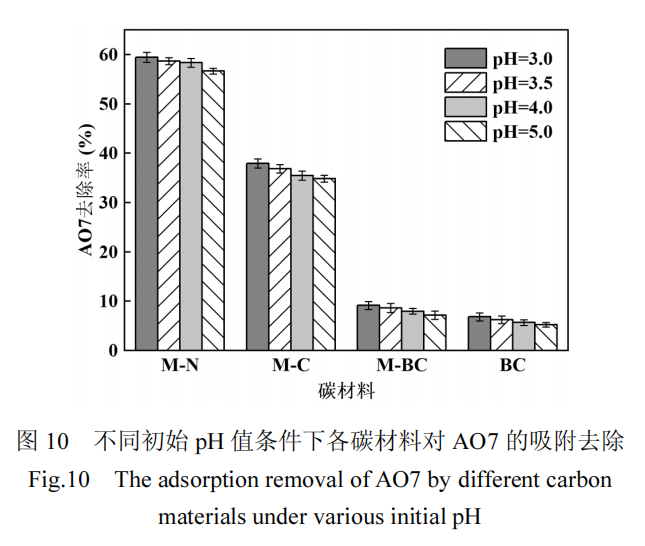
AO7 溶液的初始 pH 值为 5.0,吸附实验和 PMS引入后的催化氧化过程中的 pH 值变化如表 5 所示.由于 AO7 的 pKa 值为 1,在 pH 值变化范围内(3.0~6.0),AO7主要以带负电荷的磺酸基团的阴离子形式存在,PMS 引入后引起的 pH 值变化不会影响其形态 [6].为评估PMS引入后pH值变化对碳材料吸附效果的影响,测定了不同初始 pH 值条件下(3.0、3.5 和4.0),各碳材料在吸附平衡时对 AO7 的去除率,如图10 所示.实验结果表明,当 pH 值降低至 3.0~4.0 时,各碳材料对 AO7 的吸附效果变化不明显,说明 PMS的加入对吸附过程的影响不显著.
在加入 PMS 后,AO7 的去除率变化曲线如图11(a)所示.单独使用 PMS 对 AO7 的降解效率低,表明 PMS 自活化能力弱,强调了催化剂在活化 PMS 以增强其氧化效率中的必要性.为定量评估不同催化剂的活性,本研究采用拟一级反应动力学方程(式(17))对实验数据进行拟合,如表 6 所示.各碳材料的饱和吸附容量(qe)与反应速率常数的对数(ln kapp)呈正相关关系(图 11(b)),表明吸附和催化过程存在明显的协同效应.含氧酸盐在熔融盐体系中的引入不仅改善了碳材料的吸附性能,也显著提高了催化活性.具体而言,M-N 的 kapp分别是 M-C 的 5.10 倍、M-BC 的 22.64 倍以及 BC 的 33.48 倍,凸显了其在催化活性方面的优势.相较其它样品,M-N 的优异催化活性可归因于其增加的二维片层结构比例、更发达的孔道结构、增强的石墨化转变,以及氮掺杂引入的显著结构缺陷.二维片层结构有助于充分暴露催化活性位点;发达的孔结构促进了有机污染物的物理吸附;增强的石墨化转变提高了 M-N 中 sp²杂化碳结构的比例,从而增强了其电子传导能力;结构缺陷促进了有机污染物和 PMS 的吸附

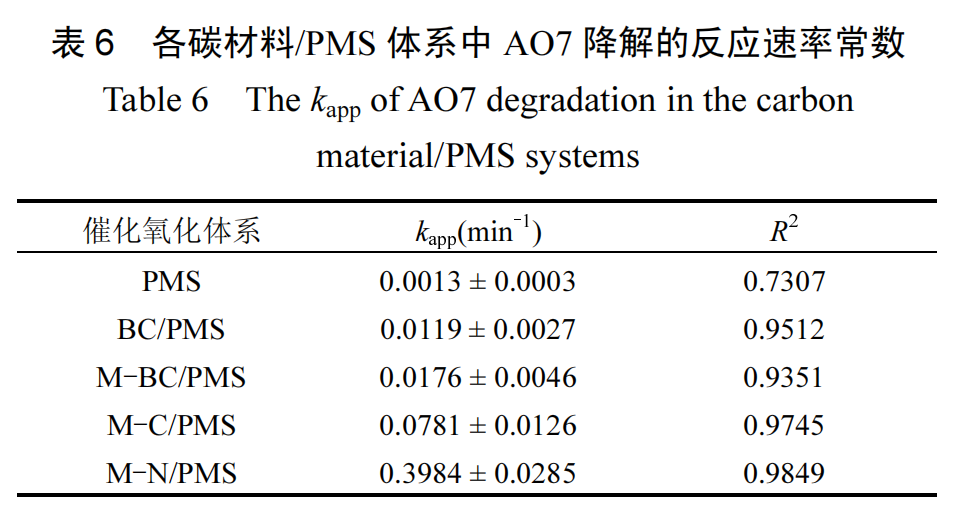
除了上述特性外,M-N 最显著的差异在于杂原子氮的掺杂.为了明确其对催化活性的贡献,本研究通过密度泛函理论(DFT)计算了不同含氮官能团以及碳空位、锯齿型/扶手椅型边缘缺陷等对 PMS 吸附的影响.使用 Materials Studio 2019 软件中的Dmol3 模块进行无自旋限制的 DFT 计算,其中交换相关函数采用了广义梯度近似(GGA)和 Perdew-Burke-Ernzerhof(PBE), 基集选择双数值加极化(DNP).能量收敛容差设定为10-5Hartree(1Hartree =27.21eV), 最大允许力和位移阈值分别设为0.002Hartree/Å 和 0.005Å.模拟使用了 H 原子钝化处理悬空键的石墨烯簇模型.
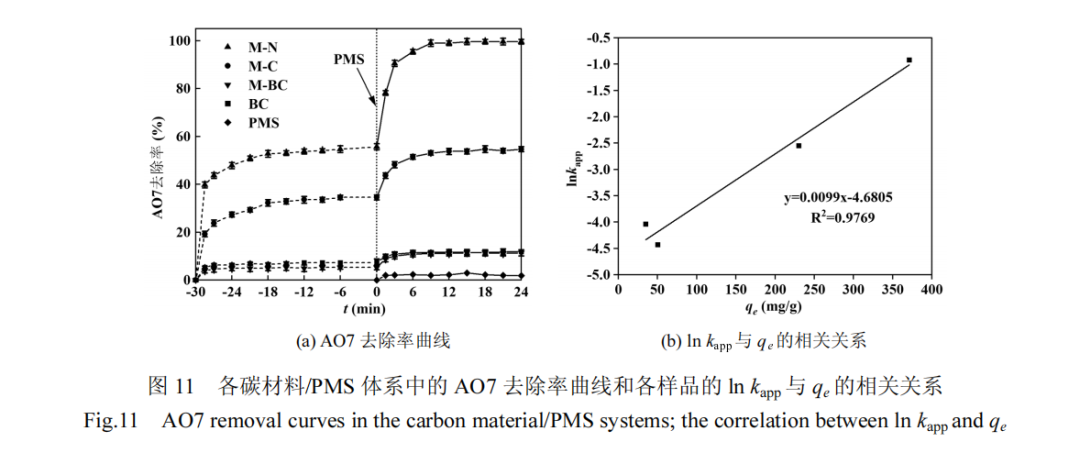
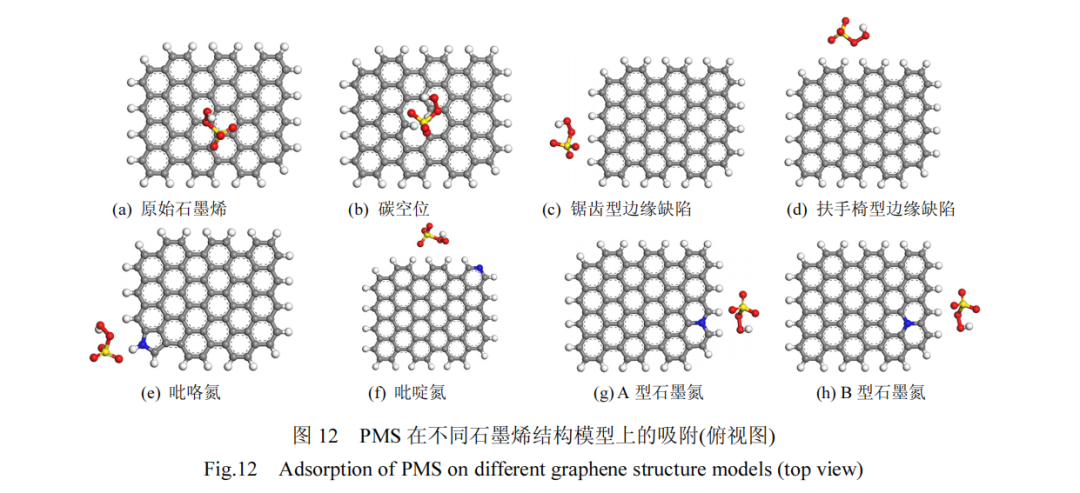
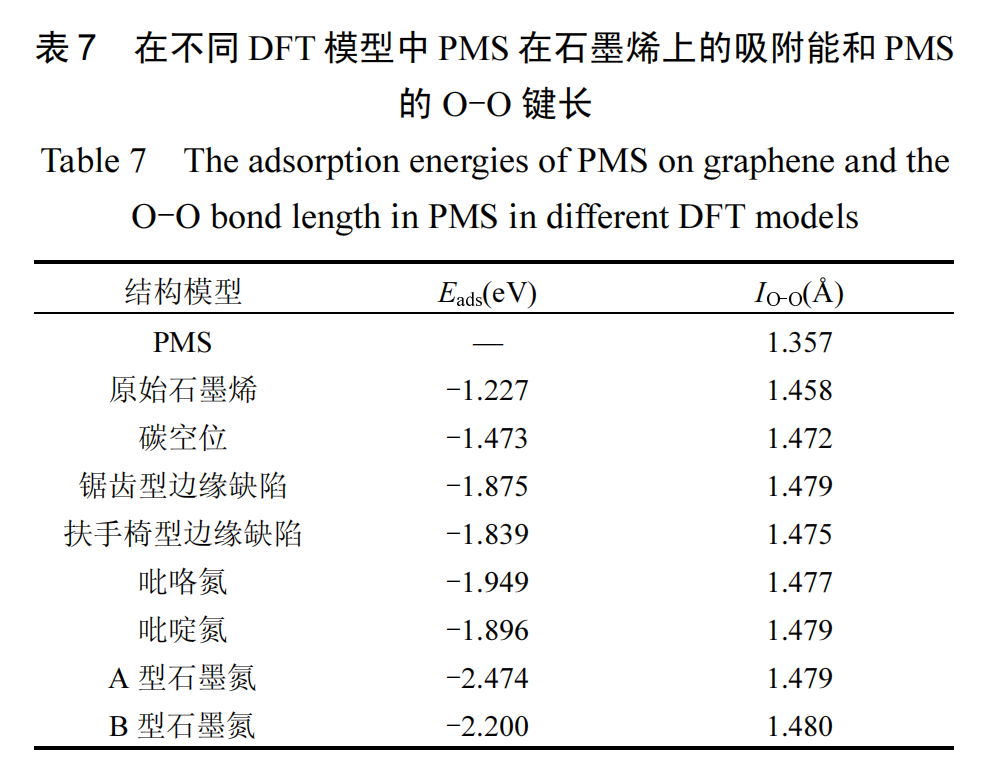
图12 展示了构建的原始石墨烯以及具有不同缺陷的石墨烯结构模型,这些类型包括碳空位、锯齿型边缘缺陷、扶手椅型边缘缺陷、吡咯氮、吡啶氮、A型石墨氮和 B 型石墨氮.进一步,本研究计算了 PMS在这些不同模型上的吸附能(Eads)及 PMS 中 O-O 键的键长变化.其中 Eads的计算依据式(18)进行:
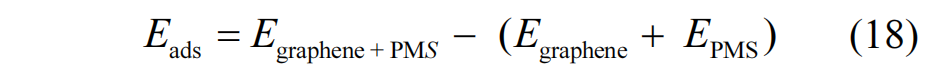

计算结果见表 7,在游离状态下,PMS 中过氧键(O-O)的初始键长为 1.357Å.接近不同石墨烯时,PMS的 O-O 键长显著增加.此外,PMS 在吡咯氮、吡啶氮、A 型石墨氮和 B 型石墨氮模型上的吸附能均超过了PMS 与未掺杂石墨烯基面结合时的吸附能,也高于PMS 在碳空位和边缘缺陷模型上的吸附能.这说明与碳空位和边缘缺陷相比,通过在石墨烯基材料中掺入杂原子氮,尤其是石墨氮,更能提高 PMS 的吸附效率,促进 O-O 键的断裂,从而有效活化 PMS.
2.4 催化机理研究
2.4.1 ROSs 的确定
本研究采用自由基淬灭实验和 EPR 测试,探究了不同含氧酸盐/熔融盐体系下制备的碳材料活化 PMS 的机理及其与材料结构间的关系.采用高浓度甲醇作为羟基自由基(⋅OH)和硫酸根自由基(SO4·− )的淬灭剂,高浓度叔丁醇为⋅OH的淬灭剂[41].如图13(a)和(c)所示,对M-N/PMS体系而言,甲醇的加入对 AO7 的降解几乎无影响,而叔丁醇的加入对 AO7 降解的抑制作用略强于甲醇.这一现象可能源于叔丁醇的介电常数(17.93)显著低于甲醇的介电常数(33.00),导致叔丁醇具有更高的疏水性[42],从而通过淬灭 M-N 催化剂的表面结合自由基,抑制AO7 的降解反应.基于以上发现,进一步采用二甲基亚砜来淬灭表面结合自由基[43].实验结果显示,10mmol/L 的二甲基亚砜显著抑制了 AO7 的降解过程,这表明在 M-N/PMS 体系中,存在大量的表面结合自由基,而游离的⋅OH 和SO4·− 几乎不产生.非自由基路径中单线态氧(1O2)对 AO7 降解的贡献通过添加糠醇来体现,结果发现糠醇使 M-N/PMS 体系中AO7 降解的 kapp 降低了 51.39%.由于糠醇还可有效淬灭⋅OH 和SO4·−[44],且这两种自由基对 AO7 降解的贡献已被排除,因此确认 1O2 是促使 AO7 去除的重要活性组分.此外,加入对苯醌对超氧自由基( O2·− )淬灭后得到的kapp与糠醇处理后的结果接近,考虑到对苯醌也可与 1O2 快速反应[45],因此仅通过淬灭实验不能确认 O2·− 在 M-N/PMS 体系内存在.
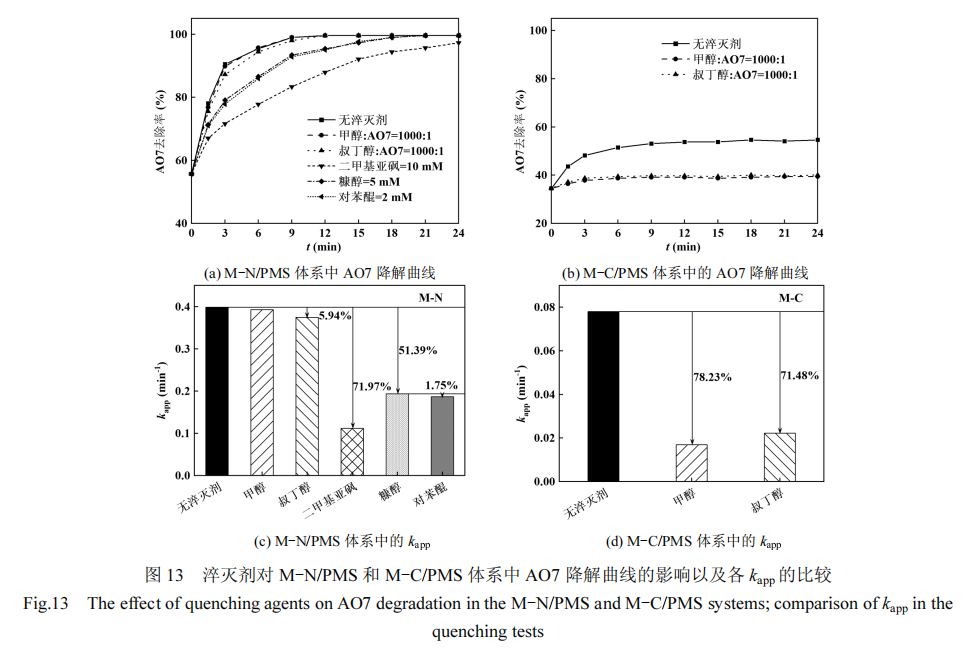
如图 13(b)和(d)所示,与 M-N/PMS 体系不同,在M-C/PMS 体系中加入高浓度甲醇和叔丁醇后,对AO7 的降解反应产生了显著的抑制效果,其 kapp 分别降低了78.23%和71.48%.这表明在M-C/PMS体系中,主要是通过⋅OH,以及少量的SO4·− 来实现AO7的降解.
由于淬灭剂可能会在催化剂表面与 PMS 或AO7 之间发生竞争吸附,这种相互作用可能会对淬灭剂的抑制效果产生放大影响[13].此外,淬灭剂对多种 ROSs 均具有清除能力[44].因此,仅用自由基淬灭实验确定特定类型的 ROSs 的存在并不充分,需结合EPR 测试以增强验证的严谨性.
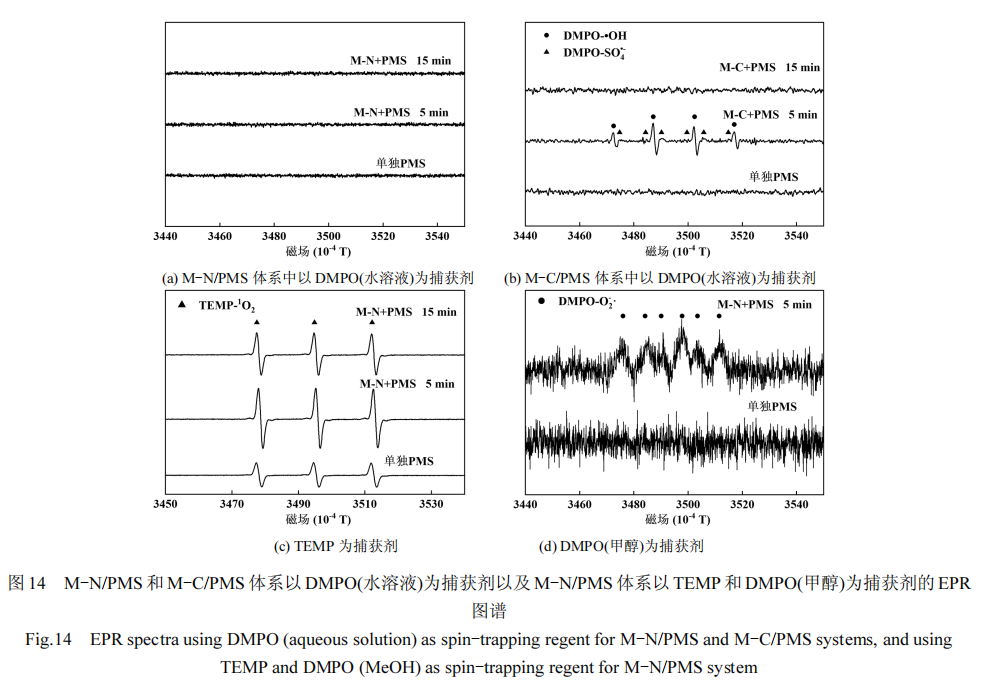
首先采用 DMPO 作为自由基捕获剂,分别在M-N/PMS和M-C/PMS体系内进行⋅OH和SO4·− 的捕获实验.如图 14(a)(b)所示,在 M-N/PMS 体系中,未检测到与 DMPO 相加和的任何自旋物质的特征信号峰.结合自由基淬灭实验的结果,可认为在该体系内几乎不产生自由基⋅OH 和SO4·− .在 M-C/PMS 体系内,反应进行 5min 时检测到了 DMPO-⋅OH 特征 1:2:2:1 四重峰和很弱的 DMPO-SO4·− 特征峰,在淬灭实验中通过甲醇和叔丁醇对 AO7 去除影响的对比中,已确认⋅OH为主要的 ROSs,而SO4·− 含量远低于⋅OH.此外, SO4·−在酸性环境下易发生自淬灭反应(式(19))[46],这进一步解释了 DMPO-SO4·− 信号强度较低.
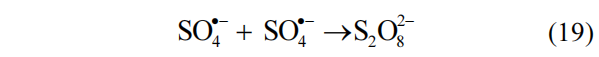
然而,当反应进行至 15min 时,体系内未再检测到DMPO-⋅OH的特征信号,表明M-C催化剂此时几乎不再产生 ROSs.这一现象与图 10 所示 M-C/PMS体系中 AO7 去除率的趋势相符.尽管 AO7 的浓度较高,其去除率随反应时间的增加不再提高,表明以自由基机制为主的 M-C 的催化活性迅速衰减.
随后,利用 TEMP 来捕获 M-N/PMS 中的 1O2,如图 14(c)所示.体系内的 TMPO-1O2 信号强度随着M-N 的加入而显著增强,结合自由基淬灭实验,证实了 M-N 在活化 PMS 过程中产生了大量的 1O2.此外,在甲醇溶液中进行的 DMPO 捕获 O2·− 的实验中,观察到如图 14(d)所示的微弱的 DMPO- O2·− 加合物信号,表明体系内存在少量的 O2·− .
因此,综合自由基淬灭实验和 EPR 测试的结果可知,M-N 催化剂活化 PMS 降解 AO7 的机制主要是通过表面结合自由基和 1O2 的作用,辅以少量的O2·− ;而 M-C/PMS 体系内则主要依靠⋅OH 和SO4·− 来降解 AO7.
2.4.2 PMS活化机理的提出
M-N/PMS体系的自由基淬灭实验表明,即使加入不同的淬灭剂,AO7 的降解反应仍能维持一定的速率,体现其并不完全依赖于 ROSs.这表明 M-N 活化 PMS 还存在其它的非自由基机制.通过先前的自由基淬灭实验,已经确认M-N 在活化 PMS 的过程中会产生大量的表面结合自由基,其实际上是通过PMS与M-N形成表面复合物 M-N-PMS*后,碳催化剂介导了吸附的 AO7 的电子向该复合物迁移,随后复合物的分解产生了表面结合的SO4·− ,然后进一步转化为表面结合的⋅OH (式(20)和式(21))[20,47].

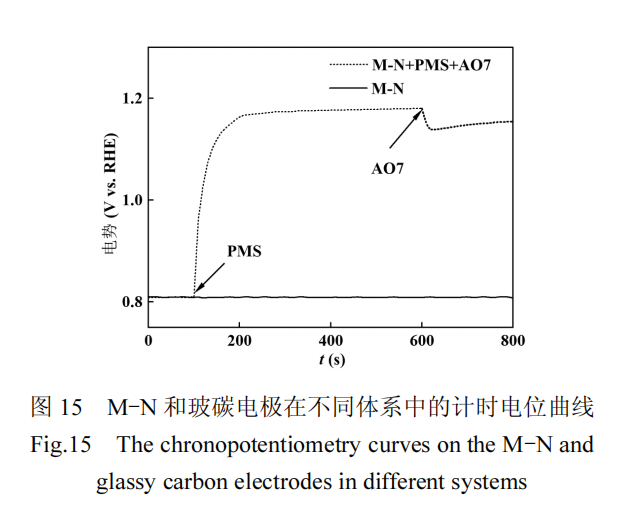
电化学测试揭示了 AO7 在 M-N 催化剂的介导下向 M-N-PMS*复合物传递电子的过程.对 M-N 进行的计时电位测试结果如图 15 所示,PMS 加入后,M-N 的电位出现增加并最终趋于稳定,这一稳定电位反映了 M-N 与 PMS 相互作用后形成的亚稳态复合物.当向该体系中引入 AO7 污染物后,观察到复合物的电位有所下降,这表明 AO7 的电子被转移到了亚稳态复合物上,导致复合物的分解及污染物AO7 的氧化降解.由此证实了在 M-N/PMS 体系中,电子传递是 AO7 降解过程中重要的非自由基途径.
DFT 计算结果显示,氮掺杂显著降低了 PMS 在其邻近碳原子上的吸附能.因此可推断 M-N 中氮的掺杂为形成大量亚稳态中间体提供了潜在的活性位点.此外,M-N 的二维掺氮结构可能促进了其电子传导性能的增强.因此,碳结构缺陷位点包括碳空位、锯齿型和扶手椅型边缘缺陷以及氮掺杂共同促进了 PMS 的吸附,PMS 或向这些缺陷位点提供电子使其被活化产生 1O2 和O2·− (式(22)-(25))[48],或与碳催化剂形成亚稳态复合中间体,与有机污染物 AO7发生电子转移并形成表面结合自由基,从而进一步降解 AO7.
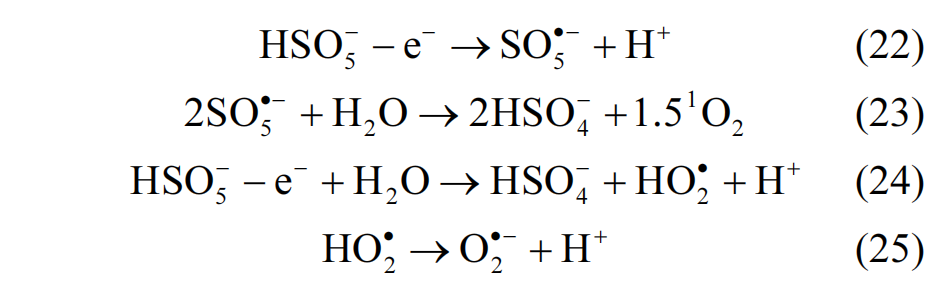
本研究通过引入含氧酸盐,有效减少了制备高石墨化和多缺陷位点的多孔碳纳米片过程中所需的熔融盐用量,从而降低了碳材料的生产成本,为大规模应用奠定了基础.所制备的 M-N 材料在吸附AO7 和活化 PMS 降解 AO7 方面表现出优异性能,证明了其在高效去除有机污染物方面的应用潜力.未来研究可进一步探索该材料在不同有机污染物治理中的应用,并结合实际工艺条件进行验证,以提升其在废水处理中的应用价值.
3 结论
3.1 含氧酸盐在熔融盐辅助葡萄糖热解体系中促进了片层结构和孔结构的形成,有助于在低熔融盐比例下合成具有高石墨化度和多缺陷位点的多孔二维碳纳米片.其中,KNO3 因其更强的氧化性和氮掺杂的引入,使 M-N 展现出更加明显的结构特征.
3.2 含氧酸盐/熔融盐体系显著提高了碳材料的吸附能力.M-N 对 AO7 的吸附动力学符合拟二级动力学模型,其最大吸附容量为 480.77mg/g.M-N 发达的孔结构、丰富的缺陷位点和 sp2 碳杂化结构增强了其对 AO7 的物理和化学吸附能力.
3.3 吸附与催化过程呈现协同效应,M-N 样品在高吸附性能的基础上表现出优异的 PMS 活化性能,AO7 在 M-N/PMS 体系中的降解反应速率常数为 0.3984min-1,分别是 M-C 的 5.10 倍、M-BC 的22.64 倍以及 BC 的 33.48 倍.结合结构表征和 DFT计算发现,M-N 的高催化活性源于发达的孔结构、高石墨化度和多缺陷位点,尤其是氮掺杂,不仅增加了结构缺陷,也是最关键的活性增强因素.
3.4 M-N 活化 PMS 以非自由基机理为主,包括形成 M-N-PMS*亚稳态复合中间体,碳催化剂介导的AO7 和 M-N-PMS*之间的电子转移,表面结合自由基和 1O2.
向上滑动阅览
参考文献








郝熔江,顾翔宇,李松庚.基于生物质基碳纳米片的水污染物氧化脱除 [J]. 中国环境科学, 2025,45(1):144-157.
Hao R J, Gu X Y, Li S G. Oxidative removal of water pollutants based on biomass-derived carbon nanosheets [J]. China Environmental Science, 2025,45(1):144-157.
(生态修复网)(转自:生态修复网)
新浪财经ESG评级中心简介
新浪财经ESG评级中心是业内首个中文ESG专业资讯和评级聚合平台,致力于宣传和推广可持续发展,责任投资,与ESG(环境、社会和公司治理)价值理念,传播ESG的企业实践行动和榜样力量,推动中国ESG事业的发展,促进中国ESG评估标准的建立和企业评级的提升。
依托ESG评级中心,新浪财经发布多只ESG创新指数,为关注企业ESG表现的投资者提供更多选择。同时,新浪财经成立中国ESG领导者组织论坛,携手中国ESG领导企业和合作伙伴,通过环境、社会和公司治理理念,推动建立适合中国时代特征的ESG评价标准体系,促进中国资产管理行业ESG投资发展。















热门推荐
抽查均符合国标!千禾味业董事长独家回应:“千禾0”就是零添加! 收起抽查均符合国标!千禾味业董事长独家回应:“千禾0”就是零添加!
- 2025年03月23日
- 16:16
- APP专享
- 扒圈小记
 4,125
4,125
土耳其爆发十多年来最严重骚乱
- 2025年03月24日
- 00:10
- APP专享
- 扒圈小记
- 1,640
日本警察厅:自4月起全面取消女警裙装制服,统一采用便于执勤的裤装制服
- 2025年03月23日
- 22:56
- APP专享
- 北京时间
- 1,327

24小时滚动播报最新的财经资讯和视频,更多粉丝福利扫描二维码关注(sinafinance)
投资研报 扫码订阅
股市直播
-

宋谈股经今天 08:17:35
今日共49股涨停,连板股总数12只,24股封板未遂,封板率为67%(不含ST股、退市股)。焦点股方面,海洋王(sz002724)、雪龙集团(sh603949)双双上演“地天板”,深海机器人(sz300024)概念股邵阳液压(sz301079)20CM3连板。短线行情依旧低迷,全市场超30股跌停,其中襄阳轴承(sz000678)、国脉科技(sz002093)、福鞍股份(sh603315)、奇精机械(sh603677)等人气股2连跌停。 -

徐善武今天 07:24:17
今天暴力杀跌微盘股,低空经济机器人(sz300024)大跌,中证1000到了60日线支撑位,短线有个超跌反弹,盘后继续探底,做好应对。现在是低位补涨不过夜,高位补跌止不住!做什么都不赚钱。 恒生科技,今天强势震荡整理,没有大跌,但是已经跌破了30日线支撑,到了颈线支撑位,具体上午有分析,可观望一下。 中证白酒,到了半年线支撑位附近,macd绿柱初现,短线还有回调,观望。 医疗与生物医药,距离下方60日线比较近了,可观望也可以适当低吸布局。 光伏与新能源车,杀机暴露,涨的少跌幅大,观望。 半导体与科创50,目前到了60日线支撑位,不可再杀跌了,观望或者低吸。 通信设备与cpo,接近前期支撑位,观望。 计算机人工智能大数据云计算软件等,有点跌破60日线,有点接近60日线支撑,稳健的观望。 传媒与动漫游戏,马上到了60日线支撑位,稳健的观望。 中证军工,macd绿柱初现,还有下跌空间,不动。 低空经济与人形机器人(sz300024),没有止跌,今天出局有点晚,可等个反弹减仓。 -

巨丰投资张翠霞今天 07:09:10
4小时运行结束,总结全天市场运行,1)尾盘权重个股带动指数修复,红盘报收,微盘股重挫,3 月下旬是重要的过渡期,即将进入 4 月财报披露期,历史经验显示,在这一时期中小市值风格的胜率与超额收益均有所回落。市场投资者在财报季前往往会更加关注公司业绩的确定性,微盘股公司由于业绩稳定性相对较差,容易受到资金的抛售;2)量能,沪深两市今日成交额14744亿元,较上个交易日15797亿元减少1053亿元;3)行业板块方面,以加权涨幅来看56家行业13家红盘,旅游、银行、有色等板块涨幅居前;公共交通、通用机械、软件服务等板块跌幅居前;4)市场延续结构型行情,题材热点快速轮动。详细解盘,可关注《翠霞首席课》的“热点直击”和“操盘指南”~~~ -
宋谈股经今天 07:06:34
3月24日收评:沪指探底回升涨0.15%,旅游板块全天强势1、市场全天探底回升,三大指数尾盘翻红,黄白二线分化明显,微盘股指数跌逾4%。旅游板块全天强势,张家界(sz000430)、峨眉山A(sz000888)、大连圣亚(sh600593)、天府文旅(sz000558)涨停。海洋经济概念持续活跃,大连重工(sz002204)、宝色股份(sz300402)、亚星锚链(sh601890)、振华重工(sh600320)等封板。有色、磷化工等周期股走强,北方铜业(sz000737)、湖北宜化(sz000422)、六国化工(sh600470)涨停。下跌方面,脑机接口概念下挫,创新医疗(sz002173)跌停;机器人(sz300024)概念股调整,宇环数控(sz002903)等多股跌停。个股跌多涨少,沪深京三市近3900股飘绿,今日成交1.47万亿。截止收盘沪指涨0.15%,深成指涨0.07%,创业板指涨0.01%。2、板块概念方面,旅游、化肥、保险、银行等板块涨幅居前,脑机接口、算力、机器人(sz300024)、军工等板块跌幅居前。3、两市共1263只个股上涨,53只个股涨停,3789只个股下跌,48只个股跌停,24只股票炸板,炸板率35%。 -

徐小明今天 07:05:48
【盘中直播】语音课见 -

量化伏妖今天 07:04:26
【股票收评】:有止跌迹象今日市场在上周五大跌后跌幅收窄,全日大部分时间绿盘交易,市场人气低迷,盘面死气沉沉,没有像样的板块出来表演,早盘还相对有点扛劲,指数跌幅还不算大,但是下午跌幅就有点大了,14点20分跌出全日最低3340点,这时技术指标价值陡峭创下-14度的两月来最低。市场开始有点恐慌,担心会不会再像上周五一样失控,就在这时突然一股力量出手拉升指数,使得指数一口气翻红。技术指标粘合线日线惯性死叉,价值陡峭在指数回升后重回低位拐头向上-9度左右。板块方面旅游酒店,化肥行业,数字阅读,钛白粉,保险,低碳冶金等涨幅靠前;赛马概念,人脑工程,MLOps概念,微盘股,全息技术,数字水印等板块跌幅靠前。总得来看今天指数呈现出惯性探底回升态势,分时图白线在上,黄线在下,两线距离较大,说明大盘股今天是有维稳指数,尤其是银行、保险股对于稳定指数贡献不小,不过中小盘科技股继续大幅下跌释放风险,获利兑现和割肉盘一同涌出,该跑的也都跑掉了,洗盘到了短期极限,大盘也有止跌迹象。 -

史月波高控盘今天 07:03:11
你赚钱亏钱他们不管的,只告诉你这些票基本面好,然后赚你的管理费 -
史月波高控盘今天 07:03:07
这个ETF,是把高现金流的股票打了一个包,和高现金流基金不一样吗,换汤不换药,之前基金经理卖基金,基金被他们玩臭了现在又打包到ETF里。【更多独家重磅股市观点请点击】 -

北京红竹今天 07:02:51
收摊了, 一会见 -

趋势领涨今天 07:02:44
=加入潜伏擒牛VIP,享四大顶级服务=【1】购买VIP自动加入私密小直播间!【2】每周3-5只超短金股调入调出服务,适合实时看盘的投资者!【3】每周一份高端内部绝密文章:包含近期布局、热点版块、指数预判!【4】每月2~3只高端中线金股服务!(VIP超短、中线个股均有涉足,让上班族也能跟上VIP节奏!)现月课7.5折,1288元!季课6.9折,3558元,续费季度更划算!新朋友可先月课体验!点网址,直接买,订购地址:【更多独家重磅股市观点请点击】【更多独家重磅股市观点请点击】