




原标题:Aβ与tau共舞,为阿尔兹海默症掀起怎样的波澜?
原创 Maxwell 神经前研

自2000年起,中国人口老龄化的程度持续加深。到2022年左右,中国65岁以上人口将破2亿关口,达到总人口的14%[10],使中国逐渐进入老龄社会。而人口老龄化不仅仅对社会转型带来挑战,更是对国家养老保障体系的一场测验。伴随年龄的增长,疾病也不断在增加。
痴呆症并非正常老龄化过程的一部分。全世界大约有5000万痴呆症患者,每年新增病例1000万。而阿尔茨海默病(Alzheimer's Disease,AD),就是其中最常见的一种痴呆症状。
2017年全国精神疾病流行病学调查显示,我国65岁及以上人群老年期痴呆患病率为5.56%,EOAD(Early On-set Alzheimer's Disease,成人早发型痴呆)的发病率也越发增高。但神经科学领域对于AD的详细病理研究仍然有许多不足。
今天的这篇文章将探讨和分享伦敦大学学院Marc Aurel Busche教授和马萨诸塞州综合医院的Bradley T. Hyman的最新研究——β淀粉样蛋白与tau蛋白缠结在AD中的协同作用。
- wikimedia commons -
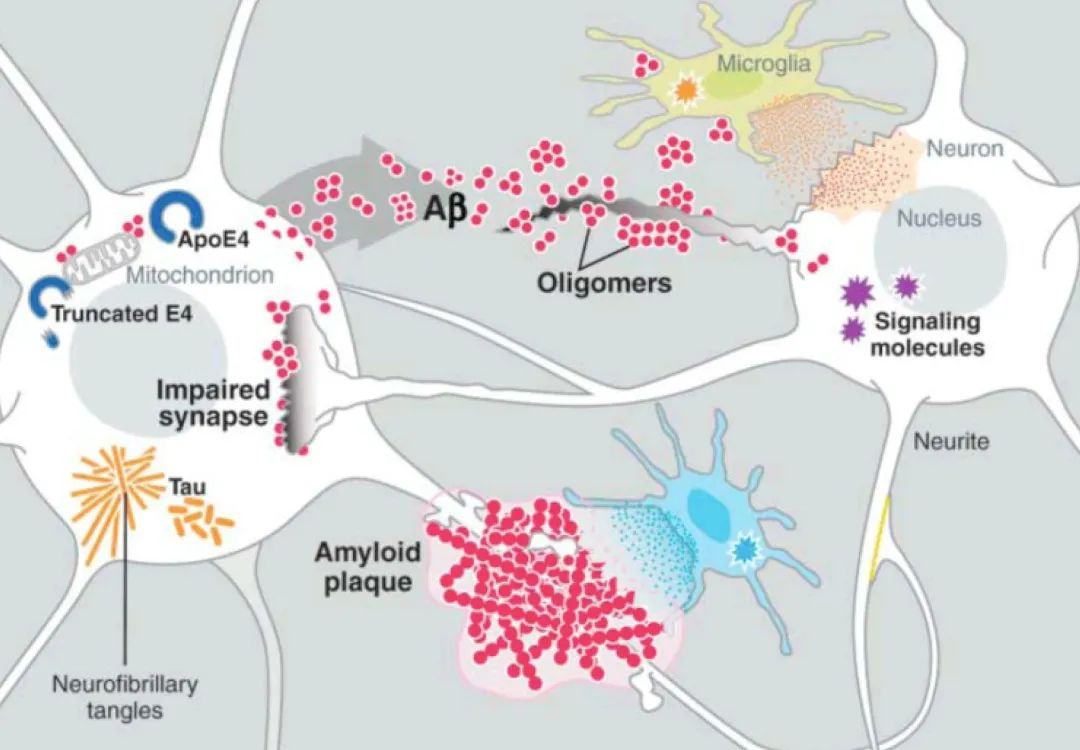
对于第一次接触到阿尔茨海默病病理过程的读者而言,β淀粉样蛋白(amyloid-β,后文将简称为Aβ)和tau蛋白是在其中绕不开的重要角色。Aβ,是一种来自神经细胞脂肪膜内的大分子蛋白质,拥有易粘性的化学特质,它是淀粉样斑块(在AD患者的大脑中发现的细胞外沉积物)的主要成分。某些类型的β淀粉样蛋白(如Aβ38/40/42)达到一定数量,会凝结起来,形成蛋白质小块,逐渐累积,便形成了斑块。这些病态的Aβ也会感染其他正常的Aβ,增加斑块的形成。而tau蛋白,是一种微管相关蛋白,它的主要功能之一是维持轴突微管的稳定性,保证脑功能的正常运作。当tau蛋白产生了缺陷,被过度磷酸化后,其结合微管的能力将会下降,在AD以及其他tau病变中所观察到的神经纤维缠结,是由过度磷酸化的tau组成的成对螺旋纤丝的集合。
所以AD的典型分子病理解释就是:Aβ形成斑块,tau蛋白形成神经纤维缠结,共同危害神经元健康。
脑皮层中广泛的Aβ斑块沉积,与皮层中tau蛋白病变所引发的神经纤维缠结,是AD最为显著的病理标志。这两者也与当今被普遍接受的AD病发机理有着密切关联。当前的AD疾病模型认为Aβ斑块的沉淀会引发一系列的链条反应,继而导致细胞内tau蛋白错误折叠和组装,继而使得病变扩散到整个神经环路以及皮层,最终导致神经系统的衰竭与认知能力的下降。
虽然有部分临床证据表明,Aβ在疾病早期的快速积累与病发年龄的提早有关。但相对于AD临床症状的进展速度,其证据更类似于偶发症状。这间接表明,AD存在一段依赖Aβ的阶段(影响病发年龄),和一段独立于Aβ的阶段(影响疾病进展速度)[22,23]。
该模型的实验支持,源自人类神经干细胞衍生的3D培养系统,及其一个AD转基因小鼠模型。该实验进一步确认了,如果Aβ斑块在tau诱导的缠结之前就已形成,那专门针对Aβ斑块的抗体只会在AD早期有效。因为早期的Aβ的降低可以减慢tau缠结的形成、传播和神经变性的速度,但此现象在后期则不会发生。
虽然AD界里普遍认为Aβ和tau在这链条反应之外没有其他相互作用。但在2019年,不同的实验和临床证据都表明Aβ的作用更为复杂。第一,Aβ的存在会增强整个AD病程中tau蛋白的表达形式与病变率;第二,Aβ和tau两者之间的相互作用在AD后期更为显著。而且这些潜在协同效应不仅能帮助阐明AD的发病机理,解释许多后期Aβ抗体实验的失败结果,还可以为tau蛋白的试验提供一条全新的研究思路[24,25]。也许减慢AD的最有效的方法是将抗Aβ和抗tau疗法相结合。在此方面更加深入的研究,也许能在不久的未来完全改变对待AD患者的治疗方式。

- Mitch Frey -
在过去几十年间,AD研究发现,Aβ斑块沉积主要聚集在新皮层的前额叶内侧和顶叶内侧区域[12]。且这些皮层斑块的聚集,在AD的临床症状出现前的10到20年内极为普遍。基于尸检和Aβ正电子扫描(PET)的研究也都表明,多达40%的认知正常个体的大脑中存在大量Aβ斑块沉积。而对于tau蛋白,研究人员发现其通常在60岁以后聚集于颞中叶。许多猜测认为,这一tau蛋白聚集现象可能先从内皮层(EC)开始,然后传播到海马体,以及边缘皮层内。这些猜测也认为,这种tau蛋白聚集现象的进展,也反映着AD患者从无症状,轻症状,到完全痴呆的进展[15-17]。
更有意思的是,研究人员发现tau蛋白的病化现象也可以独立在儿童时期出现(即在大脑的蓝斑中)。但是这一发现仍然存在许多争议,并有待更多的研究考证。但重要的是,这一系列最新的研究表明,tau蛋白的病化与扩散几乎总是与广泛的Aβ斑块沉积有关[18]。而这种现象更能启发研究人员探索Aβ斑块与tau诱发的神经缠结扩散的可能协同作用。
tau蛋白的过度磷酸化及其
传播的病理机制
在最近的一项实验里,研究人员结合了功能性磁共振和tau–Aβ正电子扫描(PET)发现,在认知正常个体的脑皮层里的Aβ,增加了神经细胞外的病态tau蛋白的扩散机率[3]。而在另一项通过张量成像来学习tau–Aβ扩散相特性的纵向研究中,其结果进一步支持了tau蛋白依赖于Aβ的传播,不是过去所猜测的相邻病理扩展机理,而是直接通过神经元连接进入脑皮层的。当实验对象变成了老年人,这一纵向项实验也发现Aβ的积累,促进了tau蛋白通过扣带束在后扣带回皮层的扩散[8]。并且,随时间的推移,在六年内,后扣带回皮层中的Aβ与tau蛋白的堆积与情节记忆的强烈下降有关。
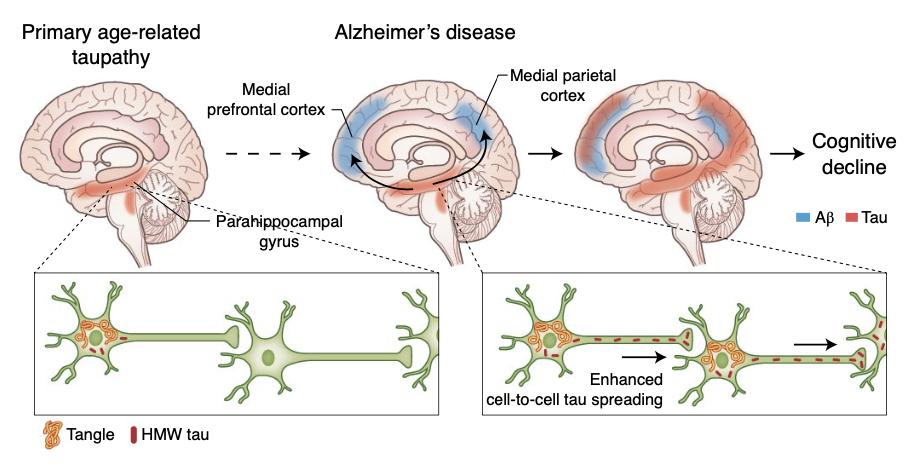
在阿尔茨海默病患者的脑内,β淀粉样蛋白所沉淀产生的斑块会逐渐加快不稳定tau蛋白的扩散,导致基本认知能力的下降*。
—
Busche et al., Nat Neuroscience
*作者注
在上半图里:当脑皮层中没有并发性斑块时,由过磷酸化的tau而导致的神经纤维缠结(红色)在许多认知能力正常的老年人的脑干核(即蓝斑核 locus coeruleus)和包含内嗅皮层(entorhinal cortex)的海马旁回(parahippocampal gyrus)内存在。而在患有AD的患者的大脑中,脑皮层斑块的产生(蓝色)与神经元tau蛋白从海马旁回向新皮层区域(包括内侧顶叶和内侧前额叶皮层)的传播有关[7,8]。
在下半图里:在只有神经纤维缠结的人类病例(左)相比,同时具有β淀粉样蛋白斑块和缠结的AD病例的高分子tau蛋白的传播和形成速度更显著。
AD是最为普遍的一种痴呆症,其所导致的认知功能下降,通常会在AD完全显现的几年前因脑葡萄糖代谢降低和区域萎缩而出现。所以可以依靠跟踪脑部氟脱氧葡萄糖(FDG)的代谢率,并通过正电子扫描仪,来检测Aβ,tau。研究人员发现,脑皮层中的Aβ和tau的组合(但不是单一的Aβ或tau蛋白)与PCC(后扣带回皮层,posterior cingulate cortex)的低代谢率有关联。这一发现可以进一步反推出Aβ和tau的协同作用对记忆力下降的影响[3]。不同实验室,也得出了相似的研究结果,表明脑皮层的低代谢率是由于Aβ-tau的相互作用而非它们的独立作用[4]。此外,通过tau和Aβ的协同作用,研究人员可以更加准确的预测个人的认知能力是正常,还是处于患AD的风险中[5]。相似的无症状或轻度AD症状个体,内嗅皮层萎缩也与Aβ和tau的协同作用密切相关[6]。
在生物信息领域越为发达的今天,研究人员也通过分析神经成像项目(ADNI)里的数据,近一步确认了只有在Aβ存在的情况下,与tau相关的脑部皮层薄化才能更有力的形成。最后研究人员从AD患者的尸检中更深一层证明,Aβ的存在能够促使一种极为罕见的易溶高分子tau蛋白的形成。而这种罕见的tau蛋白也似乎是神经细胞间传播的基质[9]。在另一项基因研究内,研究人员发现,遗传因素也似乎有着可以调节整个神经回路病理机制的能力。通过结合人类基因表达数据与来自tau蛋白和Aβ的正电子扫描结果,该实验揭示了tau和Aβ的增长与类脂物代谢基因(即APOE)相关的共享基因集有关联,特别是对于Aβ的树突相关基因和tau的轴突相关基因来说。这一结果,更进一步为Aβ和tau之间的合作提供了证据[19]。
来自多个独立且不同的AD模型实验,为Aβ-tau协同作用进一步提供了有力的支持。通过研究细胞培养基模型,研究人员发现,当在表达tau的人类细胞里添加Aβ,会导致五天后神经纤维缠结的主要成分螺旋丝(PHF)的生成。更有意思的是,缠结不仅出现在Aβ的注射部位附近,而且出现在突触连接区域。与野生小鼠相比,通过向已经表现出早期AD症状的转基因小鼠中注射人脑衍生的PHF,脑皮层tau蛋白的形成将得到进一步的增长[20]。另一项基于该实验的独立研究发现PHF的注射,加剧了tau蛋白的病化速率,尤其是Aβ斑块附近。次跟进研究巩固了又一观点:Aβ斑块提供一个有利于tau蛋白的聚集和生成的微环境,继而促进AD症状的进展[20]。
- Julia Yellow -
拮抗与协同
Aβ-tau相互作用的后果
可溶性的Aβ能导致神经元过度兴奋,削弱神经振荡,造成类癫痫活动,甚至是明显的癫痫反应[26,27]。值得注意的是,通过许多基因实验证明,许多Aβ所导致的神经元过度兴奋依靠于内源性的tau蛋白水平。因为当给予tau的遗传缺陷,这些表型都会继而消失[28,29]。
越来越多的小鼠模型基因实验也表明,tau蛋白也拥有抑制神经元的活动,并且能降低小鼠皮层内的神经元自发动作电位放电[30,31]。
在小鼠的海马体内,研究发现可溶性Aβ和tau对LTD(Long Term Depression,长时程增强)具有拮抗作用,其中Aβ降低了LTD感应的阈值,而tau则增高了LTD感应的阈值,且阻断了Aβ依赖性作用。该研究还报告了Aβ和tau对LTP的协同作用,其实验发现亚阈剂量的可溶性tau大大增强了Aβ依赖性和LTP的抑制作用[32]。
其他的一些功能性细胞的损伤,例如线粒体的吞噬以及轴突运输的损伤,在Aβ和/或tau所存在的环境也被观察到[33-35]。
最后,多项研究也表明,Aβ和tau的协同作用也导致了小鼠模型的认知能力的下降。有意思的是在另一项实验里,研究人员也发现Aβ和tau也明显导致了杆线虫出现神经和认知障碍,并缩短了其寿命[36]。
Aβ-tau协同作用的潜在机制
Aβ-tau的协同作用
当前的研究数据表明,Aβ的协同作用对tau的沉淀是一种有渗透性的现象。在小鼠模型发现,外细胞可溶Aβ能够在斑块和纤维缠结产生之前就开始在小鼠大脑中扩散。这一研究在转基因的AD小鼠中发现,也在野生小鼠中被确认[37]。
Aβ也可以诱导tau低聚物的形成[38]。而且当噬菌斑和可溶性Aβ同时存在时,两者可以增强PHF(paired helical filament,双螺旋丝)tau聚集体的扩散传播效率[39-40]。在无细胞试验中,发现Aβ通过交叉传播,直接促进了tau聚集[41],并通过基于肽的Aβ核心片段,抑制作用而将之抵消,但这也减少了Aβ自身的扩散反应[42]。在过去的体外研究表明,Aβ和tau具有结合和共聚集作用,并且在完整的大脑中,Aβ的存在会导致tau产生蛋白酶抗性,表明Aβ促进了tau的物理变化[43]。
在一系列的实验后,研究发现Aβ和tau靶突触均受到保护,tau蛋白无效的突触也受到保护,免受Aβ损伤[52]。有趣的是,苏氨酸-205上tau的位点特异性磷酸化提供了类似的保护作用,不仅与长期以来所有tau磷酸化都是有害的观点相反,而且还增强了tau-Aβ相互作用的概念。这些数据都似乎反对两种蛋白质在突触中有直接的物理相互作用(但或许,这也表明这些相互作用也许是基于一些在显微镜下无法观测的物质)。而其他的一些研究发现,Aβ和tau可能通过其对神经元的生理影响(即激活激酶,减少tau降解,调节兴奋性和基因表达)和神经胶质激活而间接相互作用。
小胶质细胞——贡献者和中介者
小胶质细胞表达的基因,包括TREM2,是检测AD的主要危险因素,这进一步暗示,个人的先天免疫系统会影响疾病进程。小胶质细胞激活是AD的关键神经病理学特征,临床上较良性的病例往往神经胶质细胞激活较少[44]。从过去的研究中发现,噬菌斑和早期的可溶性Aβ能够触发小胶质细胞的活化并促进炎性细胞因子(包括白介素-1β)和肿瘤因子的释放[45]。从另一方面看,tau的过度表达也会驱动小胶质细胞的活化,甚至在缠结形成之前,该现象也会发生。
不同转基因小鼠的实验数据也表明,该表型可能是小胶质细胞变化的早期结果,而小胶质细胞的改变直接影响了tau病理进展[46,47]。小胶质细胞能够摄取和分解具有扩散能力的tau[48],尽管效果不佳[49],而摄取tau可能反过来导致小胶质细胞活化。周围的小胶质细胞虽然可能不吸收tau蛋白,但在包含tau蛋白的神经元附近,这些小胶质细胞也可能与神经元形成异常的体细胞连接。这样,tau蛋白便有可能通过这些链接在细胞之间转移[50]。
但是,两个严谨的相反观点并不一定是相互排斥的:首先,被活化后的小胶质细胞可引起神经退行性变,并由于神经元损伤而导致tau过度磷酸化和聚集。而缺乏了小胶质细胞分离链受体CX3CR1的tau小鼠表现出增强的小胶质细胞活化作用,并加速了tau蛋白病理的发作和发展[51]。但是,小胶质细胞也可能中和Aβ和其他一些有毒的类tau蛋白的物质,从而延迟了病理扩散和其所导致的神经退行性变。而更进一步,因为TREM2的缺失,与斑块相关的小胶质细胞的继发性损失,也能增强tau病化的扩散。这表明TREM2可能可以指示包含tau的小胶质细胞来减轻tau病理症状。
结语
在现今的阿尔茨海默病的治疗发展中,医学界仍然普遍持守着Aβ和tau没有协同作用观念。但是,这近十年的实验数据,已经出现了令人信服的实验和临床证据。这些数据支持AD中Aβ和tau之间的共病原性有着协同作用,这种协同作用不仅表现在整个疾病的蔓延过程中,而且这一系列的协同作用从根本上也推动着整个疾病的发展。因此,从通过结合过去的失败与成功,重新评估AD的病理也许能够为临床治疗提供一个重要的突破。
但是,我们对Aβ-tau协同作用的知识仍处于起步阶段,尚有许多未知,比如不同模型内所产生的不一致病理原因,和其他病理过程(例如神经胶质细胞活化的作用)。此外,科学界不仅需要进一步了解血管变化和与衰老相关的过程,还需要探索其他的生物变量与Aβ-tau协同作用。阿尔茨海默病的病理研究虽然有了显著性的突破,但研究学者还需要解答更多未知的问题与现象。
参考文献
1.Adams, J. N., Maass, A., Harrison, T. M., Baker, S. L. & Jagust, W. J. Cortical tau deposition follows patterns of entorhinal functional connectivity in aging. eLife 8, e49132 (2019).
2.Hanseeuw, B. J. et al. Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann. Neurol. 81, 583–596 (2017).
3.Pascoal, T. A. et al. Amyloid-β and hyperphosphorylated tau synergy drives metabolic decline in preclinical Alzheimer’s disease. Mol. Psychiatry 22, 306–311 (2017)
4.Wang, L. et al. Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between β-amyloid and tauopathy. JAMA Neurol. 73, 1070–1077 (2016).
5.Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 19, 687–700 (2018).
6.Desikan, R. S. et al. Amyloid-β associated volume loss occurs only in the presence of phospho-tau. Ann. Neurol. 70, 657–661 (2011).
7.Synergy between amyloid-β and tau in Alzheimer’s disease
8.Jacobs, H. I. L. et al. Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals. Nat. Neurosci. 21, 424–431 (2018).
9.Fortea, J. et al. Cerebrospinal fluid β-amyloid and phospho-tau biomarker interactions affecting brain structure in preclinical Alzheimer disease. Ann. Neurol. 76, 223–230 (2014).
10.http://www.chinanews.com/gn/2020/06-19/9216394.shtml
11.https://www.cellsignal.cn/contents/research/tau-protein-and-neurofibrillary-tangles/tau-protein
12.Villeneuve, S. et al. Existing Pittsburgh compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain 138, 2020–2033 (2015).
13.Braak, H. & Braak, E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 18, 351–357 (1997).
14.Jansen, W. J. et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. J. Am. Med. Assoc. 313, 1924–1938 (2015).
15.Crary, J. F. et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766 (2014).
16.Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 19, 687–700 (2018).
17.Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T. & Hyman, B. T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639 (1992).
18.Pontecorvo, M. J. et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140, 748–763 (2017).
19.Sepulcre, J. et al. Neurogenetic contributions to amyloid beta and tau spreading in the human cortex. Nat. Med. 24, 1910–1918 (2018).
20.Ferrari, A., Hoerndli, F., Baechi, T., Nitsch, R. M. & Götz, J. beta-Amyloid induces paired helical filament-like tau filaments in tissue culture. J. Biol. Chem. 278, 40162–40168 (2003).
21.He, Z. et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 24, 29–38 (2018).
22.Choi, S. H. et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278 (2014).
23.Lee, H. K. et al. Three dimensional human neuro-spheroid model of Alzheimer’s disease based on differentiated induced pluripotent stem cells. PLoS One 11, e0163072 (2016).
24.Israel, M. A. et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220 (2012).
25.Busche, M. A. et al. tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 22, 57–64 (2019).
26.Palop, J. J. & Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 17, 777–792 (2016).
27.Zott, B. et al. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 365, 559–565 (2019).
28.Roberson, E. D. et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754 (2007).
29.Roberson, E. D. et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 31, 700–711 (2011).
30.Marinković, P. et al. In vivo imaging reveals reduced activity of neuronal circuits in a mouse tauopathy model. Brain 142, 1051–1062 (2019).
31.Menkes-Caspi, N. et al. Pathological tau disrupts ongoing network activity. Neuron 85, 959–966 (2015).
32.Ondrejcak, T. et al. Soluble tau aggregates inhibit synaptic long-term depression and amyloid β-facilitated LTD in vivo. Neurobiol. Dis. 127, 582–590 (2019).
33.Vossel, K. A. et al. tau reduction prevents Aβ-induced axonal transport deficits by blocking activation of GSK3β. J. Cell Biol. 209, 419–433 (2015).
34.Ittner, L. M. et al. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc. Natl. Acad. Sci. USA 105, 15997–16002 (2008).
35.Fang, E. F. et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412 (2019).
36.Wang, C., Saar, V., Leung, K. L., Chen, L. & Wong, G. Human amyloid β peptide and tau co-expression impairs behavior and causes specific gene expression changes in Caenorhabditis elegans. Neurobiol. Dis. 109, 88–101 (2018). Pt A.
37.Bennett, R. E. et al. Enhanced tau aggregation in the presence of amyloid β. Am. J. Pathol. 187, 1601–1612 (2017).
38.Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Guerrero-Muoz, M. J., Jackson, G. R. & Kayed, R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041 (2010).
39.Vergara, C. et al. Amyloid-β pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 137, 397–412 (2019).
40.Gomes, L. A. et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 138, 913–941 (2019).
41.Vasconcelos, B. et al. Heterotypic seeding of tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of tau-pathology in vivo. Acta Neuropathol. 131, 549–569 (2016).
42.Griner, S. L. et al. Structure-based inhibitors of amyloid beta core suggest a common interface with tau. eLife 8, e46924 (2019).
43.DeVos, S. L. et al. tau reduction in the presence of amyloid-β prevents tau pathology and neuronal death in vivo. Brain 141, 2194–2212 (2018).
44.Perez-Nievas, B. G. et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136, 2510–2526 (2013).
45.Prinz, M., Jung, S. & Priller, J. Microglia biology: one century of evolving concepts. Cell 179, 292–311 (2019).
46.Yoshiyama, Y. et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351 (2007).
47.Maphis, N. et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138, 1738–1755 (2015).
48.Asai, H. et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593 (2015).
49.Bolós, M. et al. Direct evidence of internalization of tau by microglia in vitro and in vivo. J. Alzheimers Dis. 50, 77–87 (2016).
50.Hopp, S. C. et al. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflammation 15, 269 (2018).
51.Cserép, C. et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 367, 528–537 (2020).
52.Bhaskar, K. et al. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31 (2010).
作者:Maxwell | 封面:Simon Prades



“掌”握科技鲜闻 (微信搜索techsina或扫描左侧二维码关注)











